
基于转录组异常表达构建结直肠癌特征基因预后风险评分模型
2021-04-13 05:55包汝娟陈慧芳叶幼琼
上海交通大学学报(医学版) 2021年3期
包汝娟,陈慧芳,董 宇,叶幼琼,苏 冰
上海交通大学医学院上海市免疫学研究所,上海200025
结直肠癌(colorectal cancer,CRC)是最常见的恶性肿瘤之一,发病率和死亡率均较高[1]。目前,CRC 患者的预后主要取决于诊断时的疾病进程和肿瘤分期[2]。然而,由于缺乏适当的诊断方法,预后指标并不能满足实际的临床需求[3]。因此,开发能够有效预测患者预后的评价指标十分必要。目前,以靶向程序性死亡受体-1(programmed death-1,PD-1)为代表的免疫检查点抑制剂是CRC 免疫治疗的主要研究方向,但仅有一小部分CRC 患者受益,且在其他癌症中免疫治疗也并非适合于所有患者[4-5]。因此,找到能够预测癌症患者免疫治疗效果的关键特征基因,将有助于指导癌症的临床治疗。近年来,随着微阵列技术[6]和RNA 测序技术的飞速发展,由此产生的转录组数据成为了探索疾病机制、挖掘各类疾病预后标志物的丰富来源[7-9]。因此,整合来源于不同技术平台的数据集,将会为全转录组水平分析肿瘤相关异常基因提供更综合的数据基础。
因此,本研究利用生物信息学技术整合公共数据库的CRC 数据集,对CRC 预后相关的关键基因进行筛选并构建模型,评估模型的预测性能,探索模型在预测免疫治疗效果方面的应用,为CRC 患者的预后管理和个体化精准免疫治疗提供参考。
1 资料与方法
1.1 CRC相关数据的获取及整理
RNA-seq 数据集:①从基因型-组织表达(genotypetissue expression,GTEx)数据库(https://commonfund.nih.gov/gtex)中下载308例正常结直肠组织样本的基因表达数据及对应的捐赠者临床信息(包含捐赠者的性别、组织来源等),即为GTEx 数据集,将基因表达数据中每千个碱基的转录每百万映射读取的片段数(fragments per kilobase of exon model per million mapped fragments,FPKM)值进行log2(x+0.001)转换。②在癌症基因组图谱(the cancer genome atlas,TCGA) 数据库(https://portal.gdc.cancer.gov)中下载471例CRC组织样本及41例正常结直肠组织样本的基因表达数据以及对应的患者临床信息(包含患者的年龄、性别、生存状态、生存时间等),即为TCGA 数据集,对基因表达数据的FPKM 值进行log2(x+1)转换。随后,使用sva R包的ComBat函数将GTEx 数据集和TCGA 数据集整合为一个数据集,即为RNA-seq数据集。
微阵列数据集:在基因表达综合(gene expression omnibus,GEO) 数 据 库[10](https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi)中下载10 例CRC 患者的基因表达数据及对应的患者临床信息(包含患者的年龄、性别、生存状态、生存时间等),包括GSE8671[11]、GSE18105[12]、GSE20916[13]、GSE23878[14]、GSE37364[15]、GSE21510[16]、GSE33113[17]、GSE39582[18]、GSE17536[19]和GSE17537[19]。使 用oligo R 包(http://www.bioconductor.org/packages/release/bioc/html/oligo.html)的Robust Multiarray Averaging函数对原始数据进行处理。由于前8个数据集同时包含了CRC 组织和正常结直肠组织的样本,故用于后续差异分析;后3 个数据集包含了模型研究部分所需的临床信息,故而作为模型验证集进行后续研究。
1.2 差异表达基因筛选及功能分析
使用R 语言limma 包[20]分别对每个微阵列数据集中的差异表达基因(differentially expressed genes,DEGs)进行筛选,再通过R 语言RobustRankAggreg (RRA)包[21]获得前8 个微阵列数据集中共有的DEGs。基因表达的差异用P 值和差异倍数(fold change,FC)的对数(log2FC) 表 示。将P<0.05 且|log2FC|>1 的 基 因 视 为DEGs。随后,分别对RNA-seq 数据集、TCGA 数据集中的DEGs 进行筛选,方法及筛选标准同上。使用R 语言clusterProfiler 包[22]分别对微阵列数据集、RNA-seq 数据集获得的DEGs 进行基因本体数据库(Gene Ontology,GO)功能分析,结果以P<0.01 为入选标准。采用R 语言GOplot包[23]呈现GO功能分析的结果。
1.3 预后风险评分模型的构建和性能评估
1.3.1 用于构建及评估模型的数据集说明 因建模构建需要,结合数据集的基因表达数据及临床信息中的生存状态、生存时间,从TCGA数据集中选择了438例符合上述要求的样本开展研究,其中随机抽取219例样本作为训练集,排除训练集的剩余219例样本作为内部验证集;同时,从微阵列数据集中选择GSE39582(556 例样本)、GSE17536(177 例样本)和GSE17537(55 例样本)数据集作为外部验证集。
1.3.2 预后风险评分模型的构建及分组标准 对“1.2”中已筛选获得的DEGs 进行二次筛选,获得GSE39582、TCGA 数据集这2 个数据共有的DEGs。采用R 语言survival包[24]进行单因素Cox回归分析,筛选与不良预后相关的基因[风险比(hazard ration,HR)>1,P<0.05],并将其作为构建模型的候选基因。基于训练集,通过R语言glmnet 包[25]将候选基因作为参数进行套索(LASSO)回归分析,确定最佳惩罚值,而后选择其对应的回归系数不为0 的基因作为建模基因,再行多因素Cox 回归分析,构建预后风险评分模型。风险评分计算公式为:风险评分=基因1表达量×多因素Cox回归系数1+…+基因N表达量×多因素Cox回归系数N(其中N代表基因数)。计算每例患者的风险评分,通过R 语言survival 包[24]的surv_cutpoint函数将患者划分为高风险组和低风险组。
1.3.3 在训练集中评估预后风险评分模型的性能 采用上述计算公式对训练集中每例患者的风险评分进行计算,并据此将其分为高风险组和低风险组。通过R语言ggplot2包绘制2组患者的风险评分和生存状态的分布图,以及建模基因表达量图谱。通过R语言survivalROC包[26]绘制时间依赖性受试者操作特征曲线(receiver operator characteristic curve,ROC 曲线),计算训练集中患者生存时间分别在1、2、3年的曲线下面积(area under the curve,AUC);通过R语言survminer包(https://cran.r-project.org/web/packages/survminer/index. html) 绘 制Kaplan-Meier(KM)生存曲线,计算2 组患者的生存率。根据AUC 值和组间的KM 生存率的差异,评估模型在训练集中的预测性能。同时,结合训练集中CRC 患者的年龄、性别、肿瘤分期等临床信息,采用多因素Cox 回归模型分析风险评分是否为判断CRC不良预后的独立因素。
1.3.4 在内、外部验证集中评估预后风险评分模型的性能 采用上述计算公式对内、外部验证集中每例患者的风险评分进行计算,并据此将每个验证集患者分为高风险组和低风险组。根据内、外部验证集的ROC 曲线和KM 生存曲线分析结果,评估该模型在内、外部验证集中的预测性能。
1.4 基因集富集分析
为验证GSE39582 数据集和TCGA 数据集中低风险和高风险组间的功能差异,我们利用基因集富集分析(gene set enrichment analysis,GSEA)[27]方法比较肿瘤特征基因集(hallmark gene sets)在2组间的富集度。其中,该肿瘤特征基因集来源于Molecular Signatures Database(MSigDB) 数据库(https://www.gsea-msigdb.org/gsea/msigdb/index.jsp),包含炎症反应、低氧等50 组基因集。分析输出结果为标准化富集分数(normalized enrichment score,NES),依据NES 的高低衡量基因集在高、低风险组间的富集程度,从而揭示高、低风险组间差异显著的肿瘤特征信号通路或生物过程。即在P<0.05 前提下,NES>1 代表基因集在高风险组中显著富集,且NES 越大富集程度越高;NES<-1 代表基因集在低风险组中显著富集,且|NES|越大富集程度越高。
1.5 预后风险评分模型评估免疫治疗效果的应用分析
本研究依据相关参考文献,下载2 个包含生存时间、生存状态和免疫治疗效果等临床信息的其他癌症的数据集,分别为348 例接受程序性死亡配体-1(programmed death ligand-1,PD-L1)免疫治疗的转移性尿路上皮癌患者的基因表达数据和临床信息[28]及49 例接受PD-1 免疫治疗的黑色素瘤患者的基因表达数据和临床信息[29],探究CRC预后风险评分模型在免疫治疗效果评估中的应用:①分析该模型在免疫数据集中是否成立。根据“1.3.2”中的计算公式对该2个数据集中的每例患者的风险评分进行计算,并据此将该2个数据集分别分为高风险组和低风险组。通过KM 生存曲线对2 组患者的生存率进行分析。②采用χ2检验统计2个数据集中不同组患者免疫治疗效果间的差异,及其与风险评分之间的关系。
1.6 统计学方法
采用R 软件(3.6.2 版本)对所有数据进行统计分析。定性资料以频数(百分比)表示,采用χ2检验进行比较。采用对数秩检验(Log-Rank 法)比较所有数据集中高、低风险组患者KM 生存分析间的差异。P<0.05 表示差异具有统计学意义。
2 结果
2.1 CRC数据统计及分析
经整合,RNA-seq 数据集的正常结直肠组织样本有349 例、CRC 组织样本有471 例。来自GEO 数据库的10个CRC数据集的基本信息见表1,因前8个数据集同时包含了CRC 组织和正常结直肠组织的样本,故用于后续DEGs分析。

表1 GEO数据库的10个CRC数据集的基本信息Tab 1 Basic information of ten CRC datasets from GEO database
2.2 DEGs筛选及GO功能分析
经筛选,GEO 数据库中前8 个CRC 数据集的DEGs数量如图1A 所示;经第2 次筛选后8 个CRC 数据集共有的DEGs 为962 个(上调427 个,下调535 个),其中最为显著的10 个上调、10 个下调的DEGs 如图1B 所示。GO功能分析结果(图1C)显示,上调基因主要富集在免疫细胞迁移、趋化因子介导的信号通路等,下调基因主要富集在负向调节细胞迁移等。

图1 CRC数据集中的DEGs筛选及其GO功能分析Fig 1 Screening and GO functional analysis of DEGs from the CRC datasets
RNA-seq 数据集中共筛选出1 749 个DEGs(上调800个、下调949 个)。GO 功能分析结果(图1D)显示,多数DEGs 富集在趋化因子介导的信号通路、免疫细胞迁移等。
2.3 CRC预后风险评分模型构建及其性能评价

图2 CRC预后风险评分模型的构建Fig 2 Construction of CRC prognostic risk score model
采用单因素Cox 回归模型对TCGA 和GSE39582数据集共有的1 927 个DEGs 进行分析,得到16 个与不良预后相关的基因(图2A)。以上述基因作为参数行LASSO回归分析,当logλ 达到最小值(-4.11,图2B,即黑色虚线标注所示)时,对应的参数为最佳建模参数,而此时有8个最佳建模参数的LASSO 回归系数均不为0(图2C,黑色虚线标注所示);对该8 个最佳建模基因[TIMP1(TIMP metallopeptidase inhibitor 1) 、 RGL2 (ral guanine nucleotide dissociation stimulator like 2)、 SLC39A13(solute carrier family 39 member 13)、IER5L(immediate early response 5 like)、HEYL (hes related family bHLH transcription factor with YRPW motif like)、 INHBB(inhibin subunit beta B)、DMKN(dermokine)和ELFN1(extracellular leucine rich repeat and fibronectin type Ⅲdomain containing 1)]进行多因素Cox 回归分析,最终得到由8 个特征基因构成的CRC 预后风险评分模型(图2D)。风险评分计算公式为:风险评分=TIMP1 表达值×0.495+RGL2表达值×0.946+SLC39A13表达值×(-1.517)+IER5L 表达值×0.418+HEYL 表达值×0.729+INHBB 表达值×0.315+DMKN表达值×0.218+ELFN1表达值×1.226。
通过上述计算公式,获得训练集中每例CRC 患者的风险评分,并据此将患者分为高风险组(N=32)和低风险组(N=187);不同组别的患者评分分布如图3A 所示,红色虚线表示分组的分界线。与高风险组相比,低风险组患者的不良生存状态较少(图3B),且8 个建模基因的表达量更低(图3C)。训练集的AUC 均值大于0.750(图3D);KM 分析结果显示,与低风险组相比,高风险组患者的总体生存率更低(图3E)。多因素Cox回归分析的结果显示,风险评分、年龄、肿瘤分期可以作为CRC 患者预后的独立因素(图3F)。以上结果证明,该模型在训练集中对CRC预后具有较高的预测价值。

图3 CRC预后风险评分模型在训练集中的性能评估Fig 3 Performance evaluation of CRC prognostic risk score model in the training set
随后,在内、外部验证集中评估该模型的性能。内、外部验证集的AUC 均值大于0.600(图4A~D);且该4 个验证集的KM生存分析显示,与低风险组相比,高风险组患者的总体生存率更低(图4E~H)。以上结果表明,该模型在内、外部验证集中的预测能力具有中等准确度。
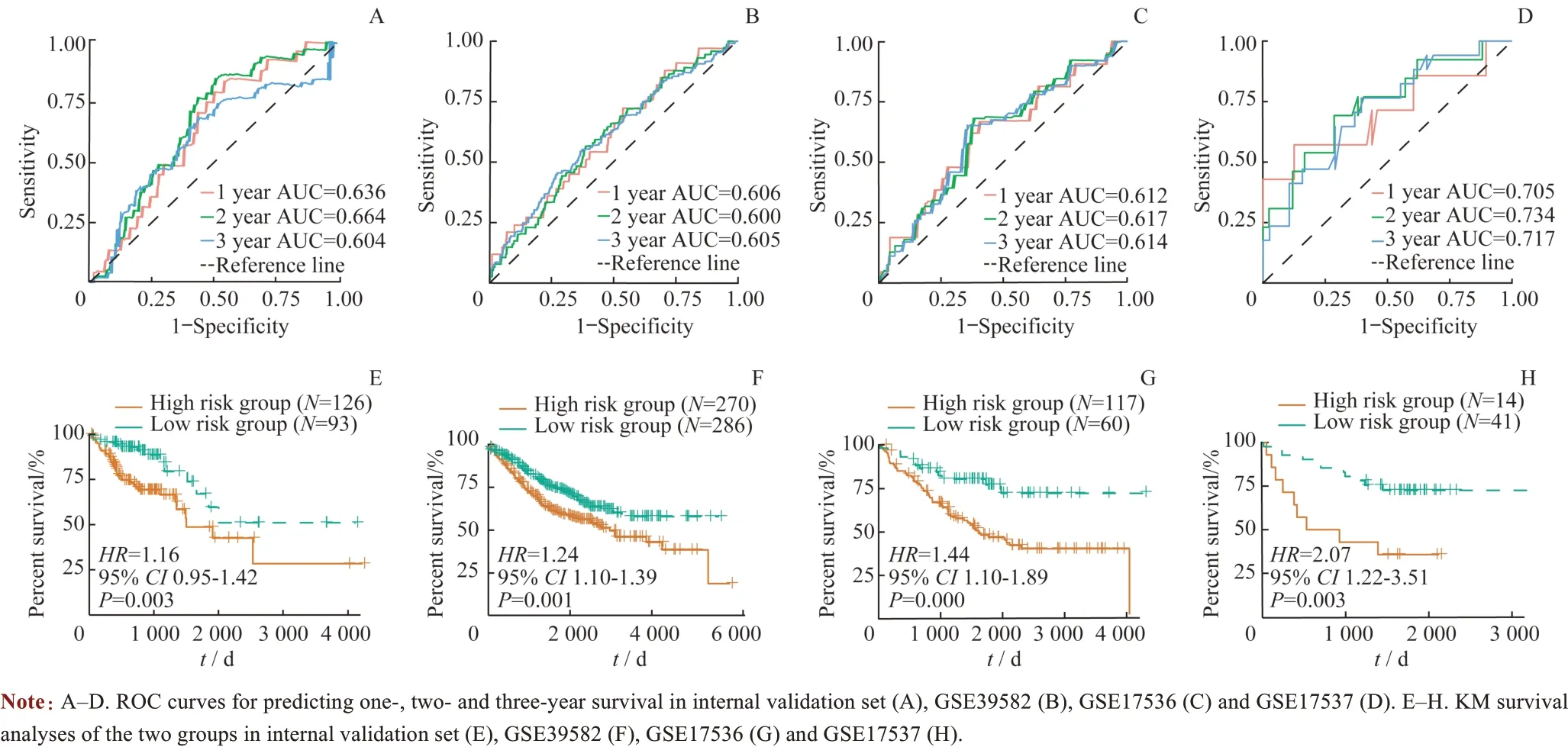
图4 CRC预后风险评分模型在内、外部独立验证集中的性能评估Fig 4 Performance evaluation of CRC prognostic risk score model in internal/external validation sets
2.4 高、低风险组肿瘤特征基因集的相关通路富集分析
GSEA 结果显示,在TCGA 数据集(图5A) 和GSE39582 数据集(图5B)中,上皮细胞-间充质转化(epithelial-mesenchymal transition,EMT)生物过程、转化生长因子β(transforming growth factor-β,TGF-β)信号通路、Kirsten 大鼠肉瘤病毒原癌基因同源产物(Kirsten rat sarcoma viral oncogene homolog,KRAS) 信号通路、炎症反应等在高风险组中具有较高的NES,而蛋白分泌、氧化磷酸化等生物过程在低风险组中具有较高的NES;从而表明,高、低风险组间的肿瘤特征基因集相关信号通路或生物过程在2个数据集中的富集均具有显著差异。

图5 TCGA数据集(A)和GSE39582数据集(B)中高、低风险组肿瘤特征基因集相关信号通路或生物过程的NESFig 5 NES of cancer hallmark signaling pathway or biological process between the high risk group and low risk group in the TCGA dataset(A)and GSE39582 dataset(B)
2.5 预后风险评分模型的应用
通过KM 生存曲线进行分别对2个数据集中高、低风险组患者进行分析,结果(图6)显示,与低风险组相比,高风险组患者的生存率均较低;继而说明,在免疫治疗数据集中风险评分的高低会影响患者最终的生存状态。因此,该CRC 预后模型在接受免疫治疗的其他癌症患者中也具有较好的评估效果。

图6 Anti-PD-L1数据集(A)和anti-PD-1数据集(B)中高、低风险组患者的生存分析Fig 6 KM survival analyses of the patients in the high and low risk group treated with anti-PD-L1(A)and anti-PD-1(B)
为了进一步分析患者的风险评分与免疫治疗效果间的相关性,排除临床信息中没有免疫治疗效果的患者后,分别对2个数据集中剩余的高、低风险组患者的免疫治疗效果进行分析,结果(表2)显示低风险组患者中免疫治疗效果为部分缓解(partial response,PR)和完全缓解(complete response,CR)的患者之和的占比较高,且经χ2检验分析显示高、低风险组患者的上述免疫治疗效果间差异均具有统计学意义(anti-PD-L1 数据集:P=0.007;anti-PD-1数据集:P=0.047)。

表2 2个数据集中高、低风险组患者的不同免疫治疗效果分析Tab 2 Analysis of immunotherapy effect between the high risk group and low risk group in the two datasets
3 讨论
由于缺乏适当的诊断方法,CRC 患者往往在晚期才能得到确诊,因此针对该疾病的临床治疗及预后研究一直是学者们关注的焦点[30]。目前,部分研究已就CRC 患者预后模型的构建进行报道,如Chen等[31]构建包含9个特征基因的CRC 预后模型,Zuo 等[32]构建6 个特征基因的CRC 预后模型,Pagès 等[33]报道含3 个特征基因的CRC预后模型,Zhao等[34]构建包含9个特征基因的CRC预后模型等。与之相比,本研究的CRC 预后风险评分模型存在如下优势:①本研究的模型构建综合了来自不同数据库的转录组数据。相比Chen 等[31]使用的3 个GEO数据集[GSE32323(癌和癌旁样本各17 例)、GSE74602(癌和癌旁样本各30 例)、GSE113513(癌和癌旁样本各14 例)],本研究使用了更为充足的GEO 样本量;相比Zuo 等[32]基于TCGA 数据集(647 例CRC 和51 例正常结直肠样本) 构建的模型,本研究不仅综合了GEO 和TCGA 的数据,还整合了正常组织样本数据库GTEx 中数据使癌(471 例)和癌旁(349 例)样本数量更为均衡。②本研究的训练集AUC 高于以往模型的训练集AUC。Chen 等[31]得出的训练集5 年AUC 为0.741,Pagès 等[32]分 析 的 训 练 集3 年AUC 为0.711、5 年AUC 为0.683,Zhao 等[34]获 得 的 训 练 集3~6 年 的AUC 分 别 为0.627、0.632、0.630、0.626,上述AUC 值均低于本研究训练集AUC 值(1 年为0.780、2 年为0.788、3 年为0.754)。同时,本研究通过3个外部验证集对预后模型的预测效能进行检验(AUC 均值>0.600),并证明该模型在其他独立数据集中也具有中等程度的准确性,而上述的前人研究并未在多个数据集中加以验证。③本研究对预后模型在预测癌症患者免疫治疗效果方面的应用做了初步探索,这是其他CRC预后模型未涉足的领域。
构建可靠的预后模型离不开对建模参数的逐步筛选,本研究整合了8 个GEO 数据集,并获得了其共有的DEGs。在显著上调的共有DEGs中,FOXQ1可在CRC 中促进肿瘤相关巨噬细胞的募集引发EMT[35];显著下调的共有DEGs 中,ABCG2 可减轻氧化应激和炎症反应,在CRC 中发挥潜在的保护作用[36]。GO 功能分析显示,微阵列和RNA-seq数据集筛选的DEGs均富集在免疫细胞迁移、趋化因子介导的信号通路等。而上述富集的生物过程或信号通路与已有文献研究相一致。如Waugh等[37]发现,在CRC 细胞中促炎趋化因子的表达较高;同时,Ackermann等[38]也证实,促炎趋化因子可促进CRC 中嗜中性粒细胞的迁移,导致免疫细胞浸润增加,且这些过程对肿瘤微环境的改变具有十分重要的作用。
本研究通过DEGs 筛选、单因素Cox 回归分析、LASSO 回归和多因素Cox 回归分析,得到8 个用于建模的特征基因,即TIMP1、RGL2、SLC39A13、IER5L、HEYL、INHBB、DMKN、ELFN1。研究[39]表明,敲低TIMP1 可抑制结肠癌细胞的增殖、迁移和侵袭,并抑制CRC 中的肿瘤发生和转移。Vigil 等[40]发现,RGL2 的异常过表达会促进胰腺癌的生长。文献[41]证实,与正常乳腺组织相比,乳腺癌组织中的SLC39A13 异常高表达,且与不良的生存状态显著相关。Ruan 等[42]发现,IER5L 在CRC 细胞中的表达较低。Liu 等[43]报道,敲低HEYL 可显著减少胃癌细胞的增殖和迁移。研究[44]发现,CRC组织中INHBB 表达可发生异常改变。Morris 等[45]对CRC患者的结肠黏膜组织进行测序,结果显示相比正常的结直肠组织,CRC 患者的结肠黏膜组织中DMKN mRNA 的非翻译区发生了异常选择性剪接和聚腺苷酸化修饰,并推测该基因可能是CRC 的新型生物标志物。Lei 等[46]报道ELFN1 可在CRC 细胞中发挥促增殖、抗凋亡和促迁移的功能。综合上述研究结果,我们发现本研究建立的模型可能与肿瘤的发生和迁移密切相关,且这些建模基因可能是肿瘤免疫微环境中发生异常改变的关键环节。
同时,本研究GSEA 结果显示高风险组患者在EMT生物过程、TGF-β 和KRAS 信号通路中具有较高的富集评分。有文献报道,EMT 通路与PD-L1 之间具有双向调节的作用[47],且该通路和PD-1/PD-L1联合靶向治疗也是一种新的免疫治疗策略[48];另有研究发现,TGF-β[49]和KRAS信号通路[50]均参与了EMT的调节。综合上述结果我们推测,高风险组患者富集评分较高的肿瘤特征通路之间可能存在相互调节,而参与这些通路的基因或许可作为高风险组患者的生物标志物。
在预后模型的应用领域,Friedlander 等[51]基于210例未接受过任何免疫治疗的黑色素瘤样本构建了包括TIMP1 在内的15 个特征基因的模型,在一定程度上可预测黑色素瘤患者的CTLA-4免疫治疗效果。本研究构建了8个特征基因的预后风险评分模型,根据风险评分将尿路上皮癌和黑色素瘤的免疫治疗患者分为高、低风险组,结果显示,高风险组患者的总体生存较差,而低风险组患者接受免疫治疗的效果更好,表现为免疫治疗效果为PR、CR的患者数量之和占比更多;上述结果表明,本研究构建的CRC 预后风险评分模型也适用于其他癌症,风险评分不同的患者的免疫治疗效果间存在较大差异,而这种差异可能影响癌症患者最终的生存率。因此,未来或可将本研究的预后风险评分模型用于癌症患者的免疫效果的预测,以节约高风险患者的时间成本,优化临床治疗方案。
综上所述,本研究成功构建了包含8 个特征基因的CRC 预后模型,对CRC 患者的预后具有一定的预测能力,可为挖掘CRC 免疫治疗新靶点、优化肿瘤免疫治疗方案提供一定的参考。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
中国现代医生(2022年19期)2022-11-04
中国现代医生(2022年19期)2022-08-25
现代临床医学(2021年4期)2021-07-31
云南医药(2021年3期)2021-07-21
家庭医药(2020年10期)2020-10-30
大众健康(2018年10期)2018-10-22
领导决策信息(2018年16期)2018-09-27
华东师范大学学报(自然科学版)(2018年3期)2018-05-14
人大建设(2017年10期)2018-01-23
