
基于温带和热带玉米群体全基因组选择和杂种优势候选位点的鉴定
2021-05-06 11:45周玲熊威胡俏强戴惠学赵涵
江苏农业科学 2021年4期
周玲 熊威 胡俏强 戴惠学 赵涵
摘要:基于187份种质资源材料的全基因组重测序数据开发了120583个高质量SNP变异位点,通过这些位点可以将具有不同遗传背景的187份玉米种质划分为两大类群,分别为包含100份材料的温带亚群和包含87份材料的热带亚群。通过对温带和热带玉米群体选择信号的遗传分化分析,检测到3664个受到选择的位点。选取187份材料中已报道能在温/热亚群形成杂种优势的135份代表性自交系,其中75份来自温带玉米自交系,60份为热带玉米系,基于两大杂种优势群进行杂种优势性状的全基因组关联分析(GWAS),[JP2]结果鉴定出2407个杂种优势候选位点不均匀分布在玉米10条染色体上。整合选择信号检测和GWAS分析结果,共识别出1153个受到选择的杂种优势相关位点,其中,619个位点与26个已报道的杂种优势相关QTLs一致。功能注释发现与候选位点紧密连锁的324个候选基因大部分都具有功能,其中包含61个重要的转录因子。根据GO富集分析发现这些候选基因主要参与了很多对杂种优势形成有贡献的关键生化代谢途径,包括氮化合物代谢、叶酸代谢、糖酵解、发育过程的负调控及转录调控等重要生物学途径。
关键词:温热带玉米;全基因组选择;杂种优势;关联分析;候选位点
中图分类号:S513.032文献标志码:A
文章编号:1002-1302(2021)04-0019-07
作者简介:周玲(1987—),女,安徽合肥人,博士,副研究员,从事玉米遗传育种学研究。E-mail:zlingxiaoyao@163.com。
通信作者:赵涵,博士,研究员,从事玉米遗传育种学研究。E-mail:zhaohan@jaas.ac.cn。
玉米在我国是重要的粮食、饲料、经济作物,尽管目前培育和审定的玉米品种很多,但是杂交种亲本趋同化严重,所用亲本自交系主要集中在少数几个骨干自交系或其衍生系,狭窄的玉米育种遗传资源已成为制约当今玉米育种的瓶颈因素,且生产上又存在病虫害大面积暴发的风险,扩增、改良和创新玉米种质资源已经迫在眉睫[1]。
杂种优势利用是玉米育种和生产的根本途径,因此在种质创新中应优先考虑创新种质的杂种优势利用问题。根据生长环境的不同,玉米可划分为温带和热带两大类群。在温带最主要的杂优利用模式是瑞德×兰卡斯特,在热带、亚热带地区则是Tuxpeno×ETO。温带种质主要表现出茎秆强韧、出籽率高、产量潜力大、农艺性状优良等特点。热带、亚热带玉米种质则表现出抗逆性强、根系发达、叶片浓绿、持绿期长等优点[2]。由于热带和亚热带玉米种质资源在病虫、干旱等生物和非生物胁迫耐受性方面具有广泛的遗传变异,[JP2]对于玉米群体改良具有重要应用价值,特别是对于温带玉米。因此利用热带、亚热带×温带杂优模式,将有利于拓宽我国温带玉米种质基础,在我国玉米育种中发挥重要作用。
由于热带玉米在经历人工驯化和改良过程中逐渐适应温带环境,因此,有必要利用选择信号检测法揭示在改良过程中发生选择的位点,挖掘候选驯化基因,进一步从基因层面上探究玉米的群体改良。例如Liu等利用温带、热带和亚热带玉米中代表性的260个玉米自交系,结合固定系数(FST)的计算方法,发现热带玉米相比于温带玉米具有更高的遗传多样性和更多的等位基因位点[3]。杨宇昕等通过对温热带玉米群体的高测序深度的SNP进行分析,在热带玉米群体和温带玉米群体中鉴定到204752个SNP标记,通过FST选择信号识别到557候选驯化基因,并鉴定到多个候选基因与玉米的开花调控密切相關[4]。[JP2]因此,利用选择信号法探究温热带玉米群体的基因组变化,可以鉴定在热带玉米适应性改良过程中受到选择的基因区段,并且深入挖掘更多与杂种优势关联的基因,对于解析杂种优势群的遗传机制和指导杂种优势群利用具有重要意义。
本研究利用来源不同的187份玉米自交系的全基因组重测序信息开发高质量的SNP位点,通过这些遗传变异位点研究自交系群体的遗传结构,划分杂种优势群,通过选择信号分析和关联分析策略主要研究以下内容:(1)鉴定温带和热带玉米在适应环境和遗传改良过程中,基因组上由于选择遗留的选择信号。(2)筛选出温热带两大杂种优势群之间的遗传变异分化关联的候选位点。(3)确定受到选择的杂种优势关联候选位点及其紧密连锁的候选基因,并分析潜在选择候选基因显著富集途径,为挖掘和利用控制杂种优势性状的位点和候选基因提供信息参考。
1材料与方法
1.1研究材料与基因组测序数据
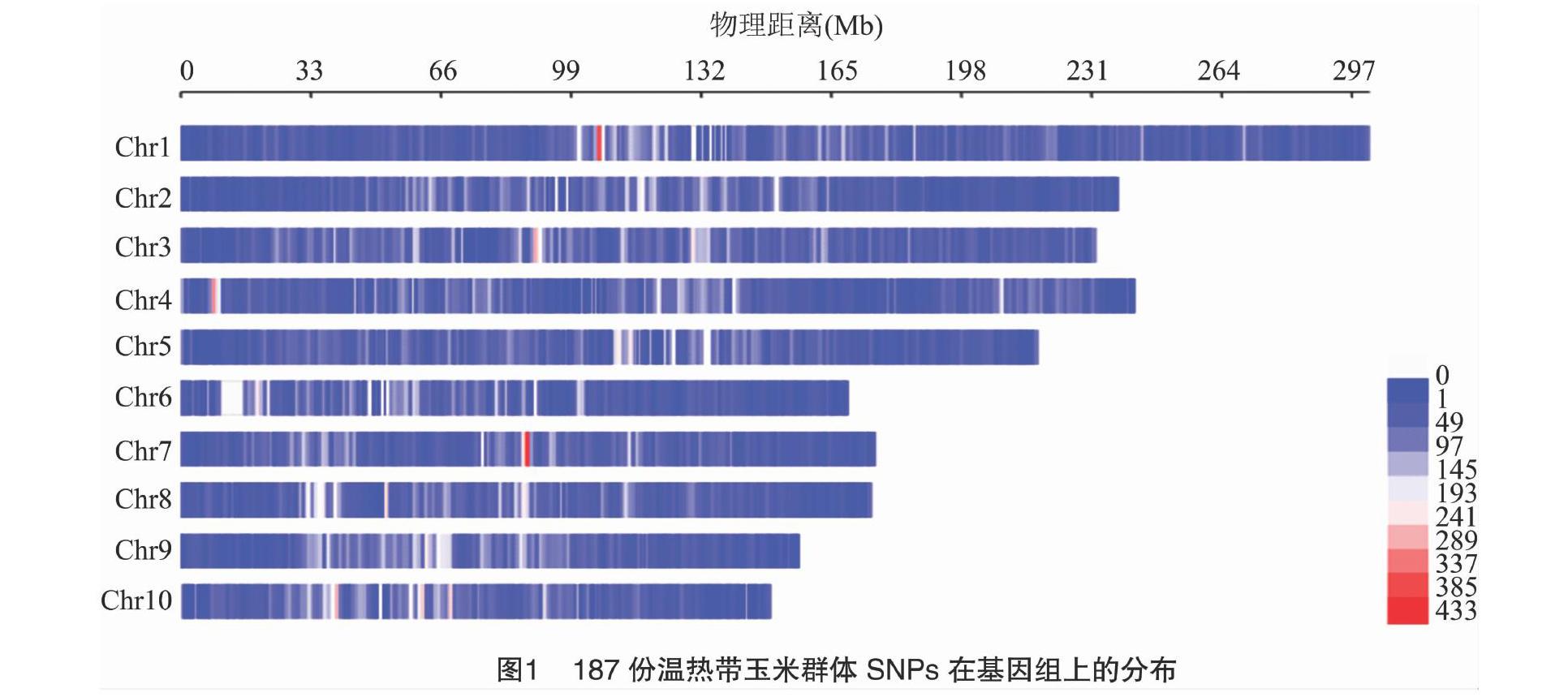
供试材料为187份玉米自交系,种质基因组重测序数据下载于NCBI网站(www.ncbi.nlm.nih.gov)的SRA数据库(PRJNA389800),对其进行过滤后利用BWA软件比对到玉米B73(v3版本)基因组上并使用SAMtool对结果进行整理,最后利用GATK软件进行SNP变异检测,获得变异位点后利用Plink软件剔除缺失率大于10%和最小等位基因频率低于0.05的SNP位点以及覆盖率较低的样本,采用Beagle软件对缺失基因型进行填补后,最终获得基因组的变异信息,SNP标记密度分布利用R软件包CMplot绘制。
1.2杂种优势类群划分
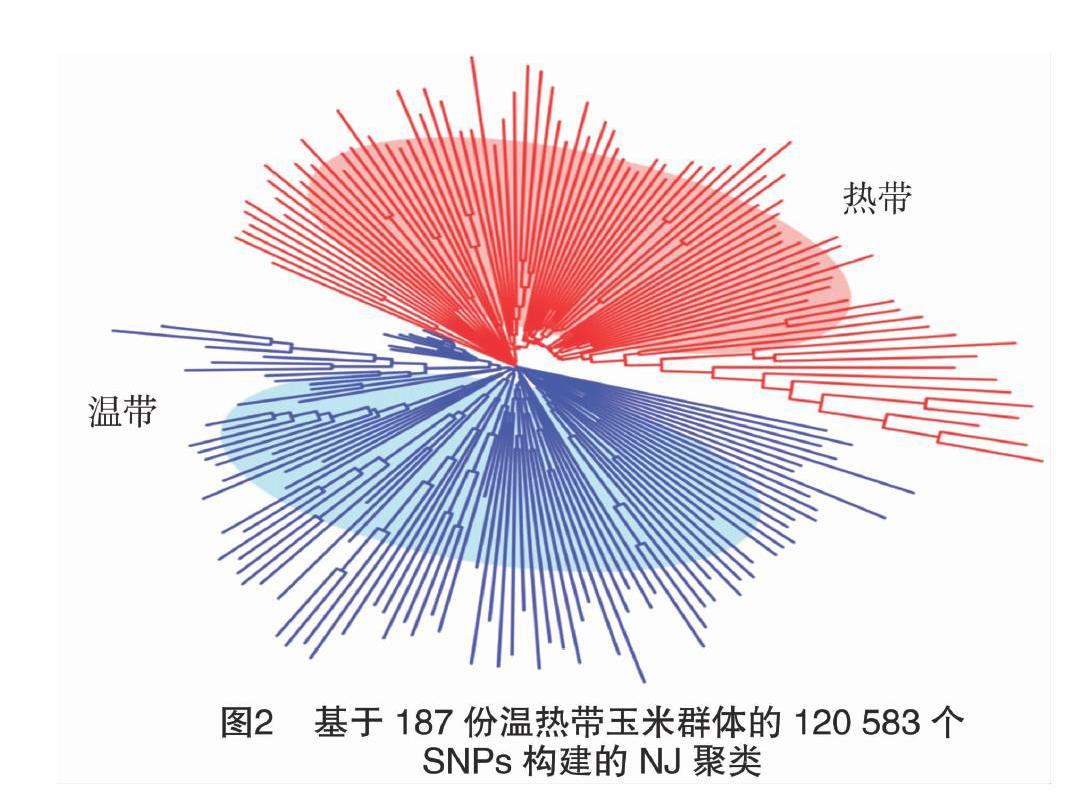
利用TASSELV5.0(www.maizegenetics.net/tassel)的邻接算法(neighbor-joiningmethod,简称NJ)[5]计算187份玉米自交系之间的遗传距离构建遗传树,并进行杂种优势类群划分。
1.3FST选择信号检测
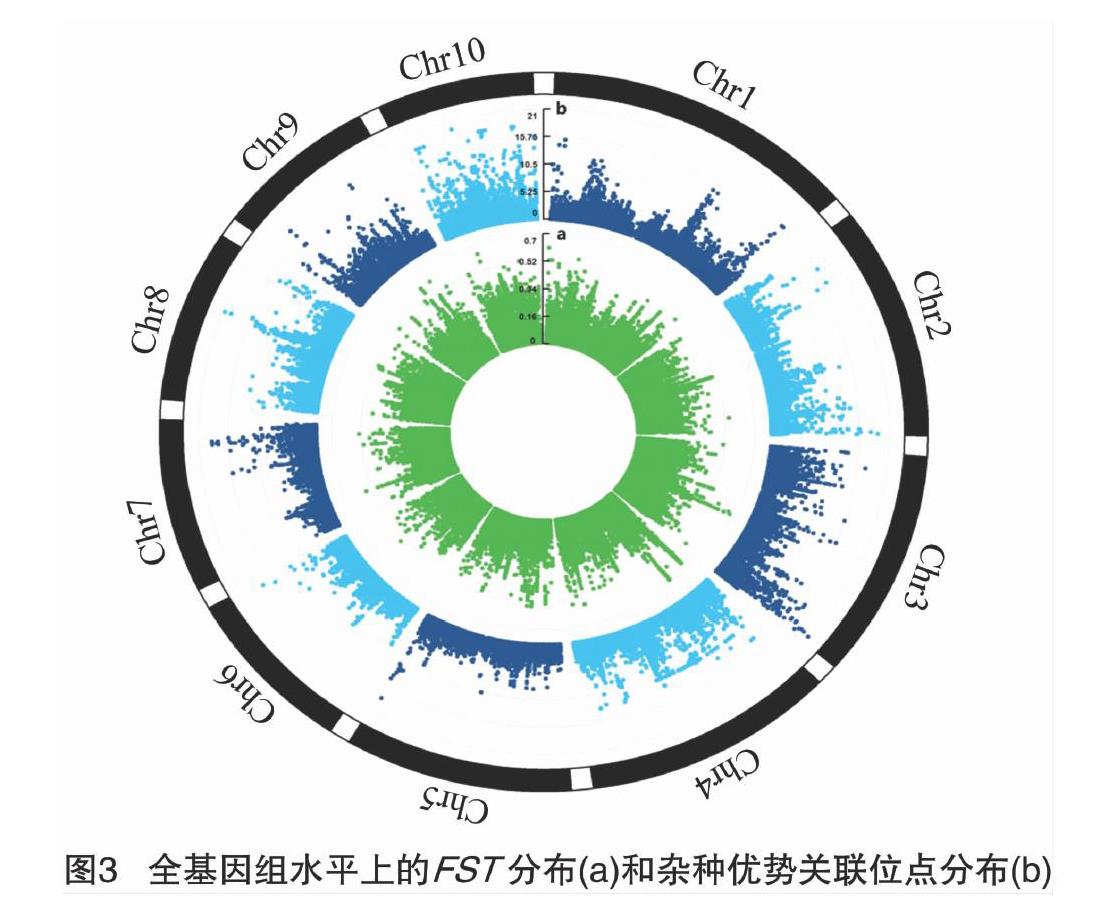
基于不同群体的遗传多样性数据,可以利用FST进行温热带玉米间选择信号的检测,其计算原理是依据染色体等位基因频率变化。在中性进化条件下,F统计量的大小主要取决于遗传漂变和迁移等因素的影响,若群体中一个等位基因因为对于特定环境的适合度较高而经历适应性选择,那么其频率的升高会增大种群分化水平,反映在F统计量上就是有较高的FST值(0≤FST≤1,FST<0.05表示种群间没有遗传分化,FST>0.25则表示种群间分化程度非常高)[6]。为了降低因单位点SNP扫描受到遗传漂变等因素影响而造成的假阳性,笔者利用VCFtools软件计算滑动窗口10kb内群体间的FST值来增加选择信号的灵敏度[7],并运用R包CMplot绘制全基因组水平上的FST结果图。为了鉴定FST值的受选择位点,选择FST值高于0.25作为显著阈值线,高于阈值线的SNP位点定义为“受选择位点”。
1.4全基因组关联分析(GWAS)
运用R软件GAPIT软件包中的压缩混合线性模型(compressedmixedlinearmodel,简称CMLM)程序[8],计算前5个主成分控制群体结构,并根据VanRaden描述的方法[9]计算亲缘关系K矩阵。在考虑研究材料群体结构和亲缘关系的情况下,对温热带两大类群进行全基因组关联分析,温带的杂种优势表型值记作1,热带记作0。采用Bonferroni方法矫正P值,鉴定温热带群体之间的特异性位点。
1.5功能注释和GO富集分析
对基于FST和GWAS共识别的特异位点进行全基因组扫描,利用SnpEffv4.3软件对变异信息进行功能注释[10],获得与其紧密连锁的候选基因,经MaizeGDB(http://www.maizegdb.org/blast.php)[JP2]数据库比对寻找有同源序列的功能基因。为了更深刻解析选择清除区域候选基因的生物学功能,利用在线平台AgriGO的单一富集分析(SEA)功能(http://systemsbiology.cau.edu.cn/agriGOv2/index.php)[11]對候选基因进行生物学过程(biologicalprocess)富集分析,筛选P值<005且错误发现率(FDR)<005的GO富集术语作为选择候选基因。
2结果与分析
2.1基因型鉴定和进化树构建
本研究利用187份种质资源材料的高通量重测序数据开发了120583个SNP位点,这些变异位点基本均匀分布于10条染色体上(图1)。采用TASSEL5.0软件的NJ法构建了系统发育树,结果(图2)显示,187份玉米材料被划分为2个不同的类群上:100份材料被聚类为温带玉米亚群,另外87份材料被聚类为热带玉米亚群。
2.2基于全基因组选择信号的检测和关联分析
基于全基因组检测到的SNP标记利用VCFtools软件计算温热带亚群体之间的FST值,结果表明,全基因组水平上FST值高于阈值线0.25的受选择位[CM(21][KG*8]点共有[KG*8]3[KG*3]664[KG*8]个(图3-a),占总变异位点数的3%。其中染色体4受到选择的显著性位点最多(913个),染色体6(188个)和染色体7(189个)最少。第4染色体46380000~46390000区间内含有最高的FST值,其值为0.66。
选取187份材料中具有广泛代表性的135份玉米自交系(表1),其中75份是选自育种程序的温带玉米自交系,代表经历显著改良并适应温带长日照环境的温带玉米(如B73、Mo17等),而60份主要来源于国际玉米小麦改良中心(CIMMYT)的热带、亚热带玉米自交系(如CML103、NC350等),代表具有丰富的遗传多样性、改良程度较低、更适应玉米驯化原始环境的热带玉米。在控制群体结构和材料间亲缘关系条件下,基于温热带两大杂种优势群体利用GAPIT软件CMLM方法对其杂种优势性状的GWAS,采用Bonferroni方法矫正P值,显著水平阈值为α=0.05/120583=4.15×10-7,在-lgP>683的水平下,共检测到2407个特异位点与杂种优势群划分显著关联(图3),其中显著的P值的范围是6.84~20.67,解释了4.31%~11.97%的表型变异。在10条染色体的分布依次为214、180、568、576、41、51、97、274、122、284个,其中在染色体3(568个)和染色体4(576个)上面存在显著富集。
整合全基因组选择信号的检测和GWAS分析的结果,发现受到选择的杂种优势相关SNPs共有1153个,其中在染色体3(233个)、染色体4(433个)和染色体10(201个)上有显著富集,在染色体1、2、5、6、7、8、9染色体上分别包含34、37、21、17、44、114、19个显著关联SNPs。与已报道的杂种优势相关QTL进行比较分析[12-15],发现619个位点存在于24个已报道的QTL区段(表2),其中染色体3中4个QTL区段覆盖了140个候选位点,染色体4中3个QTL区段覆盖了146个位点,[JP2]染色体10中3个QTL区段覆盖了178个位点,表明本研究识别的候选位点与已报道的杂种优势QTL结果高度一致。
2.3候选基因的富集分析和功能注释
利用Perl脚本和SnpEff4.3软件对受到选择的杂种优势关联的显著SNP标记与玉米B73参考基因组进行比对,并进行候选基因预测及功能注释,最终笔者获得了324个候选基因,其中包含61个转录调控功能的基因(表3)。利用AgriGO在线软件SEA对候选基因进行富集分析发现候选基因主要富集在55个GO富集术语生物过程(图4)。这些GO富集术语主要涉及到氮化合物代谢(GO:0006807)、叶酸代谢(GO:0046655)、糖酵解(GO:0006096)、发育过程的负调控(GO:0051093)和转录调控(GO:0006355)等重要生物学途径(图4)。
3结论与讨论
对玉米种质遗传多样性深入认识是合理利用种质的前提,随着分子生物学的发展,分子标记已成为作物遗传育种研究的重要手段,同时为评价玉米的遗传变异提供了方便、快捷的研究方法。利用分子标记的方法对玉米种质进行类群划分,方法易行、结果可靠,已经大规模用于玉米育种研究中。本研究以B73的参考基因组和187份玉米自交系的基因组信息为背景开发了120583个高质量的SNP分子标记均匀分布于10条染色体上,基于这些标记将187份自交系划分为2个杂种优势类群,分别为温带和热带两大类群。合理的杂种优势群,是把遗传关系较近的自交系划分为同一杂种优势群,在杂交种选育过程中避免遗传关系较近的自交系之间试组配工作,可以有效减少自交系之间的组配工作量,大大提高育种效率。
解析不同玉米类群遗传差异,鉴定与杂种优势相关的基因有助于深入了解玉米分化过程,同时为玉米种质的扩增和改良提供重要遗传资源。在本研究中,笔者结合选择信号分析和GWAS方法共鉴定出1153个受到选择的杂种优势相关候选位点,前人已报道的26个杂种优势相关QTL区段覆盖了本研究获得的619个候选位点,表明本研究的结果与已报道的杂种优势QTL结果高度一致,这为温热带种质资源的杂交和改良奠定了基础。功能注释发现与候选位点紧密连锁的324个候选基因中大部分都是具有功能的基因,利用GO富集分析发现,这些候选基因主要参与了很多对杂种优势形成有贡献的关键生化代谢途径,包括氮化合物代谢、叶酸代谢、糖酵解、发育过程的负调控及转录调控等重要生物学途径。
此外,这些候选基因中还包括了61个转录因子,在驯化过程中,植物对各种环境、组织和发育信号做出反应,需要各种功能基因的表达,从而进行精确的调控。而植物感受外界环境如干旱、病害、盐碱等体内细胞发育等信号时,需要一系列信号传递激发转录因子,从而启动功能基因的转录表达,最后通过基因产物的作用对外界信号在生理生化等方面的变化做出适当的调节反应[16]。可见,植物的转录因子通过其功能域与DNA及其他蛋白质间的相互作用,可以调节植物的生长发育及环境胁迫应答等过程[17-18]。因此,这61个转录因子将成为研究玉米驯化的候选基因。
参考文献:
[1]戴景瑞,鄂立柱.我国玉米育种科技创新问题的几点思考[J].玉米科学,2010,18(1):1-5.
[2]TallurySP,GoodmanMM.Experimentalevaluationofthepotentialoftropicalgermplasmfortemperatemaizeimprovement[J].TheoreticalandAppliedGenetics,1999,98(1):54-61.
[3]LiuK,GoodmanM,MuseS,etal.GeneticstructureanddiversityamongmaizeinbredlinesasinferredfromDNAmicrosatellites[J].Genetics,2003,165(4):2117-2128.
[4]杨宇昕,邹枨.基于温带和热带玉米群体全基因组FST和XP-EHH的选择信号检测[J].中国农业科学,2019,52(4):579-590.
[5]SaitouN,NeiM.Theneighbor-joiningmethod:anewmethodforreconstructingphylogenetictrees[J].MolecularBiologyandEvolution,1987,4(4):406-425.
[6]WrightS.Evolutionandgeneticsofpopulations[M].Chicago:UniversityofChicagoPress,1978.
[7]MaY,DingX,QanbariS,etal.Propertiesofdifferentselectionsignaturestatisticsandanewstrategyforcombiningthem[J].Heredity,2015,115(5):426-436.
[8]ZhangZ,ErsozE,LaiCQ,etal.Mixedlinearmodelapproachadaptedforgenome-wideassociationstudies[J].NatureGenetics,2010,42(4):355-360.
[9]VanRadenPM.Efficientmethodstocomputegenomicpredictions[J].JournalofDairyScience,2008,91(11):4414-4423.
[10]CingolaniP,PlattsA,WangLL,etal.Aprogramforannotatingand[HT][HT][LM]
[KH*4D]
[HT8.][KG2]predictingtheeffectsofsinglenucleotidepolymorphisms,SnpEff:SNPsinthegenomeofDrosophilamelanogasterstrainw1118;iso-2;iso-3[J].Fly,2012,6(2):80-92.
[11]TianT,LiuY,YanH,etal.AgriGOv2.0:aGOanalysistoolkitfortheagriculturalcommunity,2017update[J].NucleicAcidsResearch,2017,45(1):122-129.
[12]StuberCW,LincolnSE,WolffDW,etal.Identificationofgeneticfactorscontributingtoheterosisinahybridfromtwoelitemaizeinbredlinesusingmolecularmarkers[J].Genetics,1992,132(3):823-839.
[13]LuH,Romero-SeversonJ,BernardoR.Geneticbasisofheterosisexploredbysimplesequencerepeatmarkersinarandom-matedmaizepopulation[J].TheoreticalandAppliedGenetics,2003,107(3):494-502.
[14]FrascaroliE,CanèMA,LandiP,etal.Classicalgeneticandquantitativetraitlocianalysesofheterosisinamaizehybridbetweentwoeliteinbredlines[J].Genetics,2007,176(1):625-644.
[15][JP2]ThiemannA,FuJ,SeifertF,etal.Genome-widemeta-analysisofmaizeheterosisrevealsthepotentialroleofadditivegeneexpressionatpericentromericloci[J].BMCPlantBiology,2014,14:88.
[16]LedentV,VervoortM.Thebasichelix-loop-helixproteinfamily:comparativegenomicsandphylogeneticanalysis[J].GenomeResearch,2001,11(5):754-770.
[17]VaragonaMJ,SchmidtRJ,RaikhelNV.Nuclearlocalizationsignal(s)requiredfornucleartargetingofthemaizeregulatoryproteinOpaque-2[J].ThePlantCell,1992,4(10):1213-1227.
[18]劉欣,李云.转录因子与植物抗逆性研究进展[J].中国农学通报,2006,22(4):61-65.
猜你喜欢
蔬菜(2021年7期)2021-11-27
江苏农业科学(2016年8期)2017-02-15
江苏农业科学(2016年8期)2017-02-15
计算机应用(2016年12期)2017-01-13
软件导刊(2016年11期)2016-12-22
科技视界(2016年15期)2016-06-30
电脑知识与技术(2016年4期)2016-04-11
山西农经(2016年5期)2016-02-28
种子科技(2014年8期)2014-05-18
茶叶通讯(2014年2期)2014-02-27
