
基于深度学习的染色质交互作用预测
2021-05-13 07:16任景瑞李川张振毓邓凯
现代计算机 2021年8期
任景瑞,李川,张振毓,邓凯
(1.四川大学计算机学院,四川610065;2.四川大学网络空间安全学院,四川610065)
0 引言
染色质是由DNA、组蛋白、非组蛋白等多种物质组成的遗传物质,其结构复杂,难以直接观察,但对细胞遗传过程的基因表达有重要影响。自3C 技术问世以来,众多方法被陆续报道用于捕获染色质构象,其中Hi-C 技术是捕获染色质相互作用频次的最新最常用方法[1]。Hi-C 原始互作数据可以通过交互频次的读取序列映射到对称矩阵中,并且利用这种矩阵热图可以表示并构造为染色质的高级结构TAD[2]、隔间和染色质环等。染色质的高级结构与其功能密切相关,对基因表达和生物遗传有重要影响[3],如三维结构变化可能诱导肿瘤发育产生[4]。
目前对染色质结构的研究非常丰富,由于Hi-C 数据测量非常昂贵且耗时,但对与基因表达、转录和疾病状态相关的染色质结构有重要意义[5],所以有很多研究开始关注减少实验进行预测,对染色质结构研究存在三维结构构建,二维结构预测以及基因表达和功能性研究。对染色质三维构建目前存在多种方法,分别使用了多种距离模型算法来构建三维空间结构[6]。最新的三维结构构建方法还可以分析其基因功能[7]。而二维结构分析同样基于Hi-C 数据区域分割[8]或DNA 序列预测[9]来构建TAD、染色质环等结构,识别其区域性。染色质测序技术的发展,还对表观遗传学方面的实验应用非常重要[10],表观遗传学包括组蛋白修饰等方面,对基因表达调控和染色质重塑有重要影响[11]。分析组蛋白修饰的功能作用[12],研究染色质结构功能和基因表达[13]的影响,例如对染色质开放性[14]和染色质状态的识别[15],具有重要研究价值。
利用深度学习预测染色质交互作用能够有效获取输入数据的前后关联信息和局部特征,目前对人类细胞类型GM12878[16]和果蝇细胞类型[17]都有相关报道,使用的数据一般围绕DNA 序列和表观遗传学数据。为了深入研究组蛋白修饰和染色质交互作用两种数据相关性,本文提出了一种从组蛋白修饰数据中预测人类基因组中Hi-C 数据的方法,基于卷积神经网络,针对常见人类细胞类型IMR90 建立了深度学习模型比较预测,通过线性相关系数皮尔逊系数以及图相似性系数等评估,并最终在预测结果与原始结果之间表现出高相关性。
1 数据和方法
1.1 数据预处理
Hi-C 数据:在GEO 公开数据集上,可以通过访问代码GSE63525 下载IMR90 细胞类型的数据。我们从实验原始观测的Hi-C 序列交互数据生成原始矩阵数据,并根据分辨率确定每段基因的长度,计算对应段位置(例如本文采用10kb 分辨率,k 为一千数量,b 指代碱基段即一段碱基序列,就会将每个交互位置数据除以104,获得其bin 序号,每条染色体按细胞类型和染色体不同有上百万或上亿碱基长度)。本文关注研究染色质内交互作用,即同一序号染色体间的交互作用(同染色质间交互),最终获得22 条染色体的Hi-C 原始交互作用热图(去除性染色体影响)。
组蛋白修饰数据:从Roadmap 上可以下载每种细胞类型的所有表观遗传修饰数据,https://egg2.wustl.edu/roadmap/data/byFileType/signal/consolidated/macs2signal/foldChange/。
不同的细胞格类型对应有不同的组蛋白修饰。对于IMR90 细胞类型,可以下载以下多种修饰因素:
H3K23ac、H3K79Me1、H3K27Ac、H3K79me2、H3K27me2、H3K79me3、H3K27me3、H3K9acH3K3K36me1、H3K9me1、HP4、RPD3、H1、H3K36me2、H3K36me3、H3K9me3、H3K-4me1、H4、H3、H3、3、3me1、H4K3、3k4me1、H4K16ac
以上数据可在ENCODE 项目中公开访问,首先对组蛋白修饰的数据进行预处理,保持与Hi-C 数据同样分辨率大小(例如大小为10kb)。
1.2 实验环境
GPU:NVIDIA TU102[GeForce RTX 2080 Ti Rev.A](rev a1)
CPU:48 英特尔至强CPU E5-2650 v4@2.20GHz
内存:128GB
Python 版本和依赖包环境:Python 3.6,基于TensorFlow 的Keras。
1.3 模型与评估
本文通过多种的深度学习方法评估训练结果。训练神经网络并使其拟合的过程重,使用MSE(Mean Square Error)作为损失函数,使用MAE(Mean Absolute Error)作为目标函数。
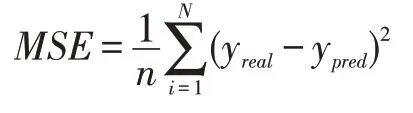
线性相关性上使用皮尔逊相关系数(PCC)和斯皮尔曼相关系数(SPCC)来评估预测结果,结果数值将显示预测结果与原始数据之间的线性相关性,相关系数的绝对值越接近1,其相关性就越强。

皮尔逊相关系数公式:
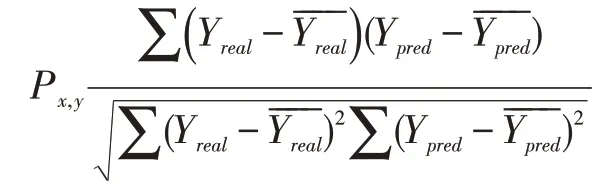
SPCC 是基于PCC 的一种相关系数计算方法,给定变量基础上给出一个排序差异集合d,由两个变量集合中的每个元素计算,最后使用PCC 公式来获取排序变量的结果。
除序列评估外,还可使用计算峰值信噪比(Peak Signal to Noise Ratio)和结构相似度指数(Structural SIMilarity)来进行评估,这两者都常用于图像处理和去噪。
PSNR 表示图像信噪比,此值越大代表失真越少,MAX=max{Ypred}-min{Ypred}。

SSIM 值的范围从0 到1,衡量两幅图的相似度,判断预测图像是否接近于原始图像:
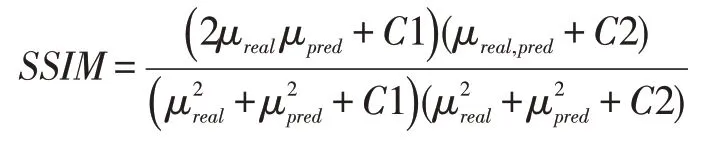
2 方法
2.1 数据预处理
对于输入数据和输出数据,本文分别使用不同方法进行预处理,因为不同数据实验测序方法不同,其表达值彼此差异较大,难以直接定性分析。
Hi-C 数据可使用标准化函数将其归一化到[0,1]的区间范围,表示每个位置交互的可能性,其原始序列交互数据可从Rao 等人(GSE63525)[18]所做的公开可用的Hi-C 实验中获得。原始数据为每个染色质每段碱基部分间的交互作用强度,在10k 碱基分辨率下,每个作用强度数据就表示染色质上按顺序排列的两段10k 长度碱基的交互频率。由于每个染色质长度彼此不同,使用字母标识i 和j 表示两个段碱基序号,它们之间的计数nij 表示染色质段上相互作用次数,通过碱基位置对应关系可构成对称矩阵,矩阵大小为N 表示染色质长度L/分辨率R(如图1 所示)。
对于组蛋白数据,首先通过bwtools 和指定bin 长度(分辨率)生成组蛋白修饰序列信号数据矩阵H,此分辨率与Hi-C 数据分辨率一致,且进行截取使实验数据长度相同,矩阵H 中每列为组蛋白修饰类型,共M列,使用最大最小值归一化使数据在[0,1]区间,每列数据表示组蛋白修饰与染色质产生作用的可能性。
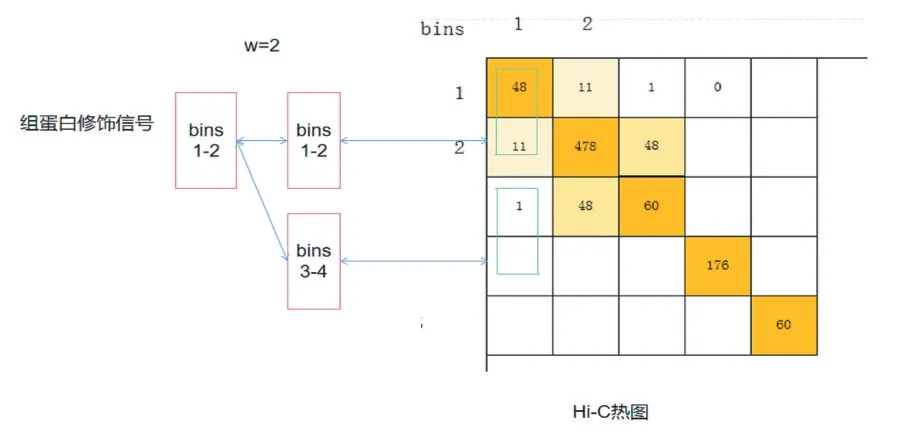
图1 Hi-C交互作用热图
2.2 Hi-C预测模型
神经网络介绍:卷积神经网络(CNN)一般用于图像处理或自然语言处理等高维特征自动提取,可学习到平移不变性等特征,在生物信息研究上也广泛应用。利用CNN 可以快速提取相应染色质交互的相关因子特征序列,构建交互作用概率分布输入。在全连接层部分,使用循环神经网络考虑染色质上下游数据带来的影响,并使所有神经元之间的参数共享,这些参数针对染色质基相互作用进行了优化,可生成用于不同组蛋白修饰的最佳滤波器,均方误差(MSE)作为损失函数,平均绝对误差(MAE)作为目标函数。对输入输出数据采用取对数值获取[-0,1]区间值域范围,并作为神经网络模型的标签和样本,可理解为对每段染色质交互作用的可能性预测。
本文提出的模型基于每对序列对应的方式,对矩阵中数据一一预测,如图3 所示,使用一个w 大小的窗口来获取每个交互基因距离下的所有数据,由于Hi-C矩阵中的对称特性,交互数据可以按列或行获取。因为染色质的交互和高维结构受到碱基段的上下游影响,有明显的区域性,对于输入的组蛋白修饰序列数据,每个交互位点的上下游各一个碱基段作为输入,共三个碱基段长度。因此,每个样本关注w 大小的染色质交互作用,使用x-1 到x+1 段(x 为交互作用发生的位置)的3 个长度的组蛋白修饰作为输入来预测相应的Hi-C 交互作用情况。本文对模型输入部分进行划分,构建一个输入模块获取两对不同位置的输入数据,此模型将在Hi-C 矩阵中的对角线区域附近生成每个bin 的交互作用数据。最后结果用热图重建方法来还原预测矩阵。窗口大小w 的选取,可使用的Hi-C 矩阵为10kb(104)分辨率下的实验数据,设定w=50,因此基因组距离为500kb,即每个碱基段包含500k(500×103)的碱基。这样可以观察交互作用密集区域,排除交互作用发生不明显的区域和较远距离的稀疏数据,使得预测结果更有价值。
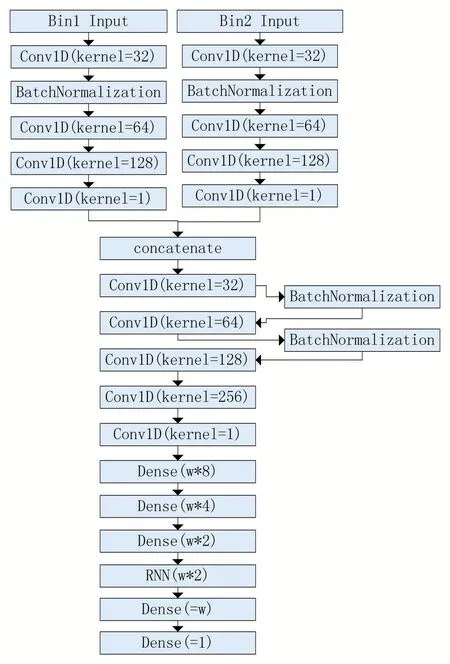
图2 染色质交互作用预测神经网络模型结构图
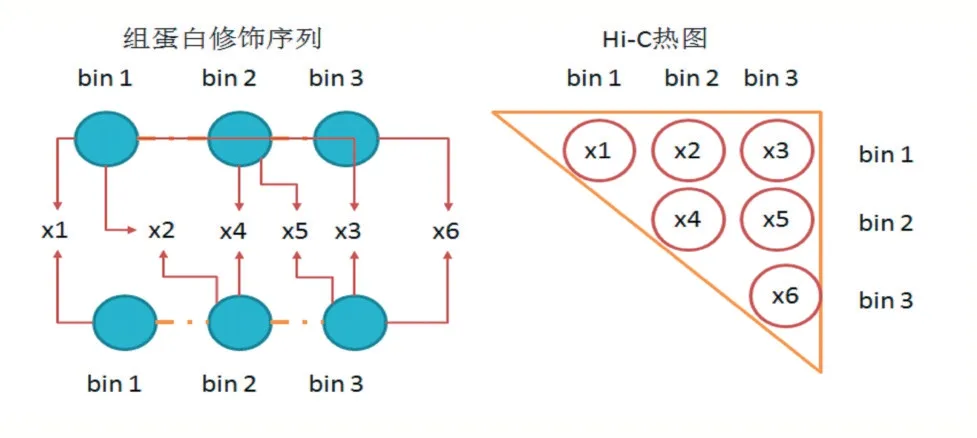
图3 两段bins输入
模型预测过程包括三个阶段,第一阶段为输入数据的卷积和特征聚合阶段,对每两对输入的组蛋白修饰信号矩阵,通过多层一维卷积神经网络获取其多种类型修饰的一维聚合特征序列,代表不同修饰类型共同作用的综合权重分布。第二阶段为拼接层,将两对输入构成二维矩阵,再次利用卷积神经网络进行不同位置间的交互作用影响特征提取。最后一层为全连接层和循环神经网络层,利用碱基上下游序列的影响,转化为时序序列的前后特征,来预测相应染色质碱基段的交互作用结果。由于神经网络预测过程中是针对各个位点进行的,需要根据位置和对称性重建矩阵结果进行对比评估,预测结果中可得到多段w 大小的预测数值排列,根据提取过程的顺序可以依次对应到交互作用发生的位置上,并使用重构算法将其恢复成数值矩阵。因此,最终结果也是对角线区域在指示基因组距离上的完整矩阵,并存在一定的拓扑结构区域。
重构矩阵热图伪代码:
M 为最后结果矩阵
For i in N:
if i <N-w+1:M[i,i:i+w]=Input[1:w];M[i:i+w,i]=M[i,i:i+w]
else:x=N-I;M[i,i:i+w]=Input[1:x];M[i:i+w,i]=M[i,i:i+w]
3 结果
我们对模型进行了多次训练,设定超参数为训练轮次设定为30 轮,批次大小每次100 个样本,优化器是RMSProp。训练完毕后可以获得一维滤波器的各项权重,表示每对固定组蛋白修饰序列的综合作用特征。最终结果分布表示序列相关性和矩阵相似性。
最终结果分别使用线性相关和图像相似性进行分析。数据集使用GEO 数据库中的GSE63525 访问代码获取的10kb 分辨率的IMR90 原始交互作用数据,重构成交互作用矩阵。其中训练集使用1-17 号染色体的Hi-C 样本,测试集使用18-22 号染色体的Hi-C 样本。最后结果显示,在500kb 的基因组距离下,对Hi-C 样本进行预测结果分析。线性相关性分析中测试集PCC 最好达到0.85,SPCC 最好达到0.8,SSIM 的测试样本中最好可达到0.98。

表1 细胞类型IMR90 染色质编号的测试数据集为染色体18 号-22 号/基因组距离(500kb)
4 结语
本文对染色质交互作用的二维结构与表观遗传学数据组蛋白修饰信号进行了相关性预测研究,提出了一种基于组蛋白修饰信号序列数据交叉预测染色质交互作用的方法。结果表明,组蛋白修饰信号在染色质相互作用中可起到重要作用,也为染色质结构预测在深度学习和不同数据上提供了一种可行路线。
对于人类细胞类型数据,具有数据量大,交互作用矩阵数据稀疏,结构作用域难以预测等特点,本文提出的方法针对稀疏数据进行了基因距离筛选,一定程度解决了数据稀疏性和不相关数据干扰的问题,并对每对数据进行分别预测,减少了不同样本差异带来的训练过拟合影响。高维染色质结构与染色质组成的内部物质高度相关,可以通过其他组成数据进行推测。
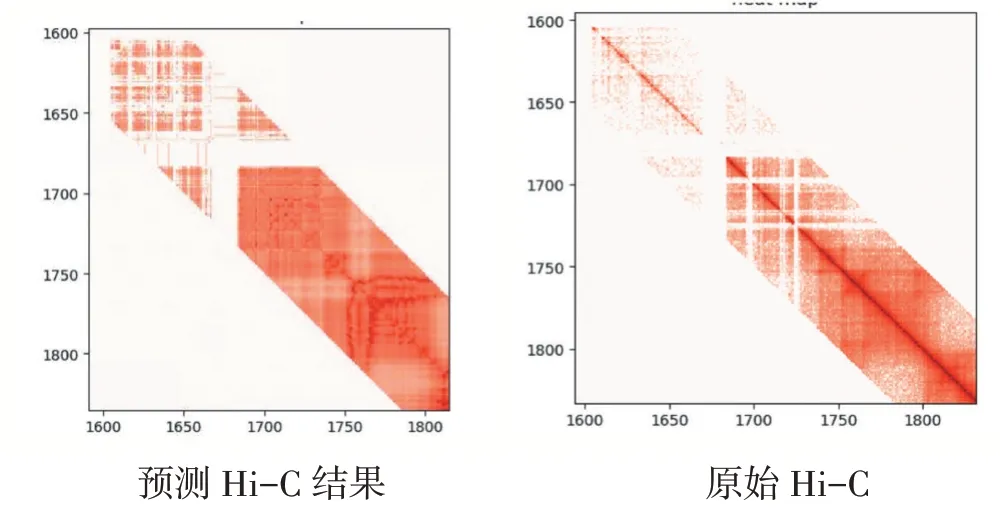
图4 chr22 16mb-18mb 上的交互
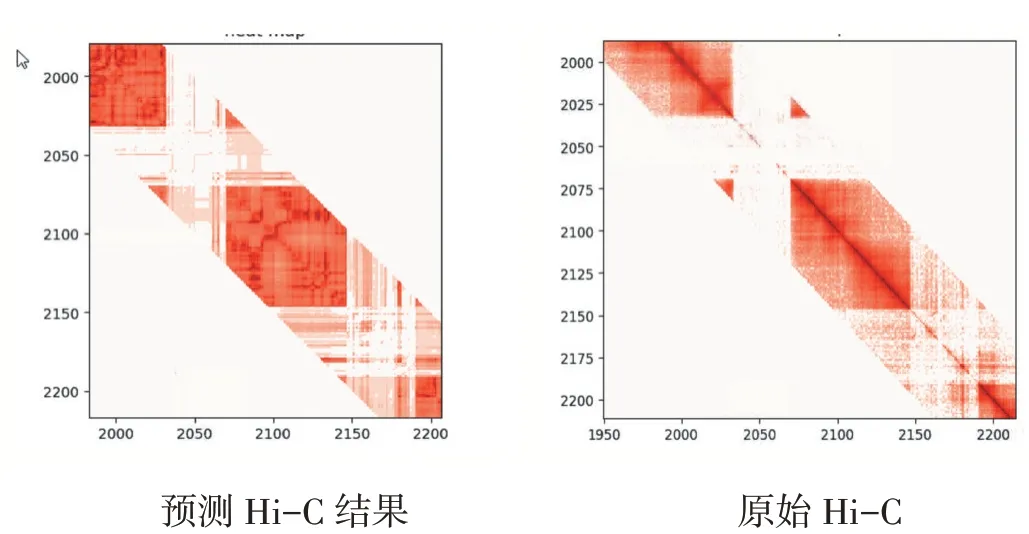
图5 chr22 20mb-22mb 上的交互
本文的深度学习模型,并应用了不同的数据集和复杂的神经网络层结构,从组蛋白修饰推测染色质数据。结果表明了模型的能力以及组蛋白修饰的方向,如何影响染色质组织。但方法使用数据类型不丰富,染色质结构不止与组蛋白修饰信号相关,输入样本数据特征仍然可以增加,模型预测数据在高维结构的留存性上仍有待提高的。
猜你喜欢
科学之谜(2021年2期)2021-04-25
河南科技(2021年35期)2021-04-25
科学导报(2020年54期)2020-09-09
学苑创造·B版(2019年5期)2019-06-14
科学24小时(2019年5期)2019-06-11
读与写·教育教学版(2017年10期)2017-11-10
CHIP新电脑(2016年3期)2016-03-10
南都周刊(2015年4期)2015-09-10
南都周刊(2015年3期)2015-09-10
南都周刊(2015年1期)2015-09-10
