
一种基于Chimera 软件的分子动力学模拟方法
2021-05-16 10:32马学婧李俊甫宋立立张兆英孙琳琳
科学技术创新 2021年13期
马学婧* 李俊甫 宋立立 张兆英 王 悦 孙琳琳
(沧州师范学院生命科学学院,河北 沧州061001)
Chimera 由美国加州大学旧金山分校的生物计算可视化和信息学中心开发,并得到了美国国立卫生研究院的部分支持[1]。作为一个可高度延伸的程序,它可应用于密度图、超分子组装、序列比对、对接结果、轨迹和构象集合,也可以生成高质量图像和动画[2-4]。Chimera 的官方网站(http://www.cgl.ucsf.edu/chimera/)上可以查阅快速入门、用户手册、命令行索引和一些教程与视频,还可以观赏用该软件制作的图片和动画。在下载页面,它还提供了Windows,Mac 和Linux 三个版本供学术、政府和非营利机构和个人使用者免费下载使用。Chimera 功能强大,界面友好,可帮助没有编程基础的教师和科研人员利用其提供的多种模块化工具进行简单的分子动力学模拟,并且可用于多种操作系统,无需购买昂贵的服务器也可以满足基本的分析需求。因此,本文将对基于Chimera 软件的分子动力学模拟方法进行阐述。
1 蛋白质结构准备
1.1 蛋白质结构可视化
首先打开一个已经下载的蛋白质结构文件(后缀为.PDB)或选择Fetch by ID,输入蛋白质结构的ID 调入蛋白质结构文件。在Presets 中根据需要选择展示模式。在Select 中,Chain 选项可以选择蛋白质复合物不同的链;Chemistry 选项可以根据化学性质,如元素、功能基团和原子轨道对蛋白质进行选择;Residue 选项可以对20 种标准氨基酸进行选择,也可以按照性质对某一类氨基酸进行选择;Structure 选项可以对蛋白质的骨架、离子、配体、核酸和特定的二级结构等进行选择。选择完毕后,可应用Actions 中的工具对选定的对象进行编辑,如:Atoms/Bonds 选项可以隐藏或显示原子或键,设置显示模式为棍型、球棍型等;Ribbon 选项可以隐藏或显示蛋白质的某一部分,还可以改变带状模式的类型;Surface 选项可以隐藏或显示蛋白质的表面,并设置显示模式和透明度;Color 选项可以对选中的对象进行上色;Label 选项可以对原子、氨基酸残基进行标注,且可自主选择标注的格式。按住鼠标左键可对结构进行旋转,按住滚轮可拖动结构改变其位置,上下滑动滚轮可对图像放大或缩小。图1 展示了Chimera 对蛋白质结构的可视化。
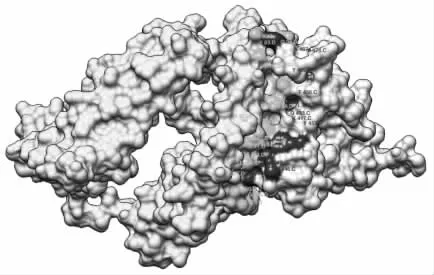
图1 蛋白质结构的可视化
1.2 突变蛋白质结构预测
对于突变蛋白的分子动力学模拟,首先需要人工对蛋白质的特定位点进行氨基酸的置换,并预测突变后的蛋白质结构。打开Tools... Sequence 工具,选择要显示序列的链,在序列上选定要突变的氨基酸,并通过Structure Editing tool 工具中的Rotamers 进行突变。选定要置换的氨基酸种类,点击Apply 之后出现多个具有实验统计可能性的结构,选择不会与其他氨基酸碰撞的最可能的结构,通常是第一个。最后通过File... Save PDB 对点突变蛋白质结构进行保存。图2 展示了突变位点的多个可能性结构。
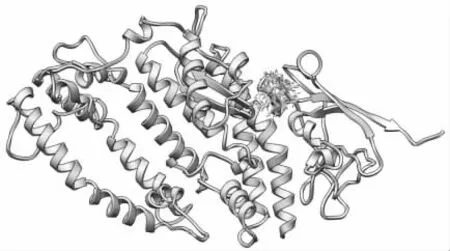
图2 突变蛋白质结构预测
1.3 蛋白质结构精炼
无论野生型蛋白质还是突变蛋白质,在进行分子动力学模拟之前需要对结构进行精炼。Chimera 使用Tools... MD/Ensemble Analysis 中的Prep Structure ... Dock Prep 工具精炼蛋白质。参数设置包括删除溶剂,替换不完整的侧链,添加氢和电荷,仅保持最高的占有率,以及将蛋氨酸/蛋氨酸/溴代-UMP转化为UMP、甲基硒代-dUMP 转化为UMP、甲基硒代-dCMP 转化为CMP。当添加氢时,每个模型都被认为与所有其它模型隔离开,并且还考虑了氢键。组氨酸,谷氨酸,天冬氨酸,赖氨酸和半胱氨酸的质子化状态是基于残基名称的。添加电荷时,标准残基根据AMBER ff14SB,其他残基根据AM1-BCC。图3 为精炼后的蛋白质结构。
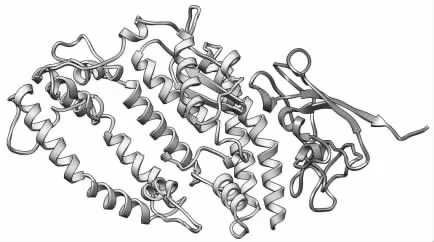
图3 蛋白质结构精炼
2 分子动力学模拟
2.1 参数设置
将所有的复合物在具有TIP3P 水的尺寸(Å)为a:10×b:10×c:10 的三斜盒中溶解,溶剂和盒子尺寸可以根据研究需要进行修改。添加适当数量的抗衡离子以中和系统的电荷。Electrostatic interaction method 和Lennard-Jones interaction method 的力场选项设置为默认值。然后,通过执行100 个最陡峭的下降步长(0.02Å),然后执行10 个共轭梯度步长(0.02Å),将复合物最小化。平衡步骤的默认设置为5000 个步长,时间步长为1 fs,步长数目和步长时间可根据研究需要进行修改,温度控制方法是Heater(298 K, 10 K/ps),输入文件保存目录,目录中不能含有中文字符。生产步骤的默认设置为5000 个步长,时间步长为1 fs,步长数目和步长时间可根据研究需要进行修改,通过耦合到Nose thermostat 保持温度,松弛时间设置为0.2 ps。通过使用Andersen barostat 将恒定压力保持在1 bar,松弛时间设置为1.5 ps,输入文件保存目录,目录中不能含有中文字符。最后点击Run,软件即按照设定的参数运行。
2.2 结果导出
模拟结束后,会自动弹出MD Movie 对话框。通过File...Save PDB, 可保存任意一帧的蛋白质结构。通过File... Record movie,可以导出模拟动画视频。均方根偏差和势能通过Analysis... Plot... RMSD 和Potential Energy 进行分析。图4 展示了分子动力学模拟后均方根偏差和势能分析图。
3 残基相互作用分析

图4 均方根偏差和势能分析
使用Cytoscape 3.6.0 的残基相互作用分析应用程序进行蛋白质结合分析。将StructureViz 下载并安装在Cytoscape 的Apps中。然后,通过StructureViz 启动Chimera。输入MMTK 格式的NetCDF 轨迹文件Production(名为prod.nc 的文件)以启动MD Movie。Analysis... Cluster 可用于基于均方根偏差的轨迹聚类,参数设置为默认值。经过计算,选择聚类结果对话框顶部的包含结构数目最多的簇用于平均结构和残基相互作用网络(RIN,residue interaction network) 的生成。点击Generate average structure for cluster 生成平均结构,保存以用于结合区分析。点击Generate residue-interaction network for cluster 生成RIN,在Cytoscape 中显示为2D 图,应用Style 中的编辑工具即可调整网络图的格式。图5 展示了残基相互作用网络图。

图5 残基相互作用网络
4 接触体积和表面积计算
打开并聚焦平均结构,使用Surface/Binding Analysis...Intersurf 进行界面显示和生成。通过Measure Volume and Area来测量界面的体积和面积。图6 展示了受体和配体的结合区域。
5 结论

图6 受体和配体的结合区域
按照上述方法进行分子动力学模拟和结果分析,可以方便快捷地对单体蛋白质、蛋白质复合物、蛋白质突变体、蛋白质截短体等进行动力学参数的测定,从而指导实验设计,导出的图片和动画还可用于教学中的课件制作,因此应用Chimera 进行分子动力学模拟,可较少地依赖硬件条件,短时间提供许多有价值的信息,服务的人群也比较广泛。现行的Chimera 版本在2018 年已经停止了继续开发,它的开发团队目前致力于ChimeraX 的研发,该软件不但具有Chimera 的所有优势,还更适用于较大的蛋白质的结构分析[5-6],将更有力地推动化学、化工、医药以及生物行业的发展。
猜你喜欢
生物化学与生物物理进展(2022年7期)2022-07-25
生物化学与生物物理进展(2022年6期)2022-07-21
小型微型计算机系统(2022年1期)2022-01-21
西安邮电大学学报(2021年1期)2021-04-19
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
无线互联科技(2020年12期)2020-09-03
科学大观园(2019年10期)2019-09-10
飞天(2019年6期)2019-07-08
中国经济周刊(2019年9期)2019-05-24
新高考·高二数学(2015年2期)2015-05-27
