
苯功能化二硫化钼纳米带输运性质的理论研究
2021-06-24 03:32胡玚玚张桂玲于海涛
黑龙江大学自然科学学报 2021年2期
纪 磊, 于 红, 胡玚玚, 张桂玲, 于海涛
(1.哈尔滨理工大学 化学与环境工程学院, 哈尔滨 150040; 2.黑龙江大学 化学化工与材料学院, 哈尔滨 150080)
0 引 言
2004年,英国曼切斯特大学的Geim团队开创性地从石墨中分离出石墨烯[1],在之后的十几年里,二维材料得到学术界的广泛关注。由于二维材料载流子的转移和热量的扩散被限制在了二维平面内,因此呈现出了许多奇特的特性。首先,由于某些二维材料的带隙可调控性,使其在场效应晶体管、储能设备和压电器件等领域应用十分广泛[2-4];另外,部分二维材料具有可控的自旋自由度与谷自由度,这使得自旋电子学与谷电子学领域的专家与学者们也对其进行了深入的研究;此外,不同的二维材料由于晶体结构的特殊性质导致了不同的电学、光学特性的各向异性(包括拉曼光谱、光致发光光谱、二阶谐波谱、光吸收谱、热导率以及电导率等性质的各向异性),使其在偏振光电器件、偏振热电器件、仿生器件和偏振光探测等领域具有很大的应用前景[5]。
过渡金属二硫化物(TMDs)是继石墨烯之后研究最广泛的二维纳米材料,它表现出与石墨相似的层状结构。其因具有优良的物理以及化学性质,而在光电、催化、储能以及传感器等领域均具有广阔的应用前景。TMDs为片层结构,垂直方向上一般仅为单个或者几个原子,而横向尺寸则可高达微米或以上级别。TMDs的范式可写为MX2,M代表过渡金属原子(如Mo或W),X代表的是硫族原子(如S或Se)。由于原子性质的相近和结构的相同,TMDs中不同的成员物理性质很相似,属性却各有不同。MoS2在所有TMDs中研究最为广泛,其优异的性质深受科研从业者的喜爱。由于其在单层时为直接带隙半导体[6],且拥有独特的电子结构和极佳的机械强度,这使得MoS2在光电器件、能量存储以及柔性电子学等领域具有重要的应用价值[7-8]。在MoS2层状结构中,S原子和Mo原子之间以共价键相连接,而MoS2层与层之间是通过范德华力相互结合的。石墨烯和MoS2之间的一个区别是是否有带隙。石墨烯没有带隙,因此只能通过应用纳米结构以及不同的化学和物理技术来引入,这大大限制了它的应用范围。而MoS2本身具有带隙,随着MoS2层数的减少,其电子性质也发生了显著的变化。带隙能从1.29 eV(多层MoS2)增加到1.9 eV(单层MoS2),带隙随层数的减少由间接带隙变为直接带隙[9]。这赋予了MoS2更加丰富的光电性质,近年来在电子和光电子器件领域展现出广阔的应用前景[10-12]。此外,带隙能量的变化以及通过改变MoS2层厚度而引起的带隙性质的变化,使少层MoS2薄膜作为吸收太阳能的光伏材料也极具优势。
基于MoS2独特的结构以及电学特性,对它的性质进行调控、拓宽其应用范围是目前的研究热点之一。具有可变电子波段的MoS2结构允许构建各种各样的异质结和超晶格,同时MoS2优良的能带排列以及可调控的载流子迁移速率,为研发高性能的电子器件与光电器件提供了优良的基础材料。近年来,各种物理和化学的功能化策略相继被提出,如插层、构建异质结构、掺杂、合金化、减小尺寸、构造缺陷、门控、光照和弯曲应变等[13]。其中较为常用的策略是,选用具有优良性质的物质插入双层MoS2夹层空间中构建新型复合插层材料。目前,Br2、H2SO4、FeCl3和碱金属等均已被证实可以通过气相插层的方式进入MoS2层间[14];此外,通过液相插层的方法,也可以成功将零价原子、分子、离子等多种粒子插层到MoS2层间;利用剥离技术已经成功将各种客体物质如Co、Ni和碱金属,插层到MoS2双层空间中[15-19]。研究结果表明,这些分子、离子或基团显著改变了MoS2材料的输运性质,使该复合材料可广泛应用于电学领域。随着对石墨烯研究的逐步深入,采用碳纳米结构对层状材料进行功能化,在优化材料性能方面取得了可喜的进展[20]。虽然插层功能化在过去的研究中已经屡见不鲜,但是苯分子对二维MoS2的交错结构纳米带进行插层功能化还未有报道。考虑到苯分子独特的结构和优异的电学性质,以及其在材料科学中广阔的应用前景,本文采用苯分子对MoS2纳米带进行插层功能化,优化其模型结构,并对其电子结构和输运性质进行理论研究。
1 模型及计算细节
主要选取三个模型对MoS2纳米带进行理论研究。双层MoS2基本结构是双层AB型堆叠方式,即两层MoS2在水平的方向上交错排列,如图1(a)所示。每个超胞中含有108个原子,其中包括36个钼原子和72个硫原子。该结构在x方向上设置了2 nm的真空缓冲区,这确保了相邻周期模块之间的解耦作用。在原有模型的基础上,对双层MoS2进行苯分子插层功能化。由于苯分子在MoS2层间可能存在

表1 MoS2、MoS2@αC6H6和MoS2@βC6H6的结合能
不同构型,所以本文对晶胞进行优化处理,让所有原子都可以在三维方向上自由运动,直到赫尔曼费曼力小于0.2 eV·nm-1。结果表明,有两种构型最为稳定,分别是在层间横向加入苯环分子形成双MoS2AB叠加结构MoS2@αC6H6,以及在层间纵向加入苯环分子形成双MoS2AB叠加结构MoS2@βC6H6,如图1(b)和图1(c)所示。对于MoS2@αC6H6和MoS2@βC6H6,每个超胞中含有120个原子,其中包括36个钼原子、72个硫原子、6个氢原子和6个氧原子,该结构依然在x方向上设置了2 nm的真空缓冲区。为了进一步证明体系的合理性,分别计算了三个体系的结合能。表1给出了三种结构结合能,其是由体系总能量减去体系各部分能量和得到的。计算结果表明,三个结构的结合能均为负值,体系为稳定结构。基于以上三个模型分别计算了用于电子结构计算的中心散射区(Bulk)体系和用于输运性质计算的双探针(Device)体系。
在Device体系中,使用金电极构建了如图1所示的双探针器件模型。在优化后,选取MoS2扶手椅方向作为输运方向,将双层MoS2交错构型作为中心散射区。为保证中心散射区足够长,设置输运方向的MoS2纳米带长度为2.305 nm,目的是分离左右电极,让左右电极之间相互作用可以被忽略。MoS2和金电极之间的距离通过金硫键(Au-S)来调控,界面处金硫键长度为0.238 nm,这个长度是金硫之间形成共价键的键长。另外,Bulk结构的晶格参数和层间距如表2所示。
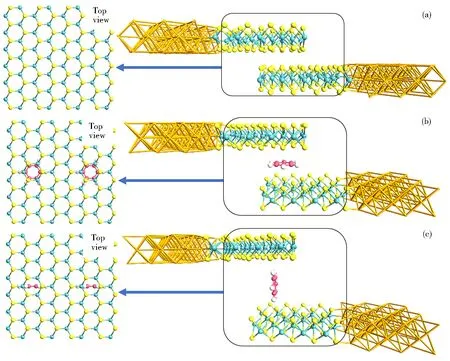
图1 MoS2 (a)、MoS2@αC6H6 (b)和MoS2@βC6H6 (c)的Bulk结构和Device结构
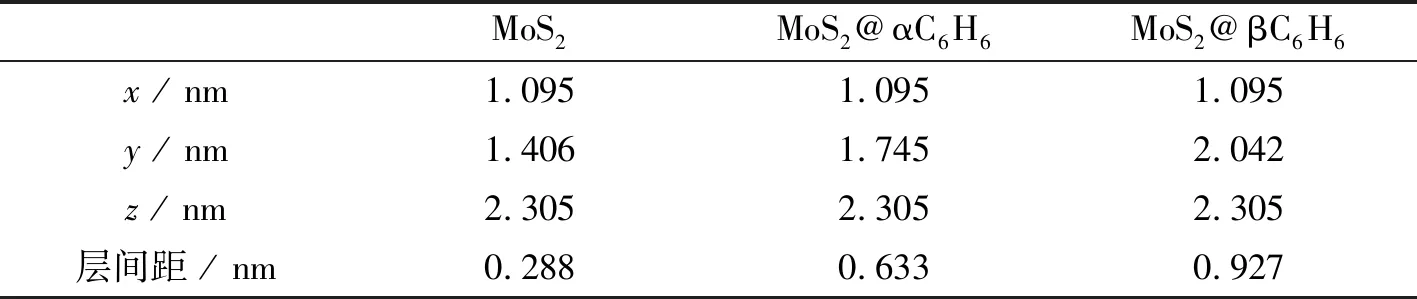
表2 MoS2、MoS2@αC6H6和MoS2@βC6H6稳定结构的晶格参数
三个器件的电子结构和输运性质是基于密度泛函理论(DFT)和非平衡格林函数(NEGF)相结合的理论原理计算得出的,运用的软件包是以从头算为理论基础的ATK软件包。在计算过程中,电子交换关系用PBE泛函中的GGA近似方法来描述,体系中所有原子的轨道基组均采用DZP。在对周期性插层体系MoS2@αC6H6和MoS2@βC6H6进行电子结构计算和晶胞优化时,布里渊区的k点值被设置为7×1×1(x×y×z)。对于计算输运性质的加电极双探针器件结构,中心散射区与电极的k点值均设置为1×1×50(x×y×z)。在计算过程中,实空间截断能与体系温度为150 Ry和300 K。在偏压为-1.0~1.0 V内,每隔0.2 V为一步计算出在各个偏压下的电流值大小。
2 结果与讨论
2.1 电子结构
图2(a)给出了MoS2的Kohn-Sham轨道、能带以及投影态密度图(PDOS)。在没有插入苯环的情况下,原始MoS2纳米带在能量为0 eV附近呈现直接带隙的半导体性质,且由PDOS可以看出,价带以及导带主要是由Mo原子的4d轨道贡献的。此外,S原子的3p电子也有少量贡献。由图2中的Kohn-Sham轨道可以看出,MoS2的带Ⅰ和带Ⅱ主要分布于两纳米带非重叠区域,而带Ⅲ则倾向分布于重叠区域。
图2(b)和图2(c)给出了MoS2@αC6H6以及MoS2@βC6H6的Kohn-Sham轨道、能带以及PDOS。当苯环分子插入二维MoS2层间时,出现了跨越0 eV的能级,插层体系的带隙消失,呈现出一定的金属性质,主要贡献该能级的依然是Mo原子的4d电子态,另外,S原子的3p电子与C原子的2p电子也对0 eV附近的能级形成有少量的贡献。但是,MoS2@αC6H6以及MoS2@βC6H6的Kohn-Sham轨道呈现出不同的分布方式。对于MoS2@αC6H6,由图2(b)可知,带Ⅰ主要分布于两纳米带非重叠部分以及苯环上,纳米带重叠区域下层的纳米带的电子云对其有一定的贡献,上层纳米带重叠部分完全没贡献。相反,带Ⅱ则主要由纳米带交错部分以及苯环贡献。对于MoS2@βC6H6,由图2(c)可以看到,带Ⅰ和带Ⅱ主要由整个纳米带结构以及苯环贡献,带Ⅲ的能带则由纳米带结构贡献,苯环对其基本没有贡献。

图2 MoS2 (a)、MoS2@αC6H6 (b)和MoS2@βC6H6 (c)的Kohn-Sham轨道、能带结构和投影态密度图
2.2 输运性质
在优化后Bulk结构的基础上,以纳米带为中心散射区,Au电极作为左、右电极,构建了扶手椅型MoS2、MoS2@αC6H6以及MoS2@βC6H6的Device分子器件,如图1所示。正偏压对应从体系的左电极流向右电极的电流,而负偏压对应从体系的右电极到达左电极的电流,本文计算所得三种结构的I-V特性曲线如图3所示。插入苯环后,MoS2双层纳米带的电流有所提高,且纵向插入苯环时大于横向插入的情况,即电流大小依次是MoS2@βC6H6> MoS2@αC6H6> MoS2。由Kohn-Sham轨道图可知,当将苯分子插层到MoS2纳米带中,纳米带的输运性质受到苯分子的影响,苯分子在层间起到类量子点的作用,参与到层间载流子的传输过程中。在苯分子的电子云参与到电子传输的情况下,量子隧穿的能量势垒高度会降低,从而导致输运能力会增强,这可以与能带结构由原来的半导体性转变为金属性相对应。

图3 MoS2、MoS2@αC6H6和MoS2@βC6H6的I-V特性曲线图
为了进一步了解苯插层功能化MoS2对输运性质影响的产生原理,计算了三种双探针结构在偏置电压为1 V时的透射光谱,如图4所示。其中偏压窗口指的是[ -V/2,V/2]的偏压范围,在图中用虚线表示。众所周知,只有出现在偏置窗口内的有效透射峰才有助于形成电流,并且较大的透射峰面积对应较高的电流值。由图4可以看到,在偏压窗口中,插层后的MoS2纳米带峰面积明显大于原始MoS2纳米带,说明电荷载流子发生了有效的传输,使插层体系具有较大的电流。MoS2@βC6H6的透射峰面积较MoS2@αC6H6略大,因此MoS2@βC6H6的电流值大于MoS2@αC6H6。综上所述,苯分子的插层功能化增强了MoS2纳米带输运载流子的能力,这与MoS2、MoS2@αC6H6以及MoS2@βC6H6的I-V特性曲线是一致的。
3 结 论
对双层交错MoS2纳米带、其苯分子横向穿插结构MoS2@αC6H6以及苯分子纵向穿插结构MoS2@βC6H6进行了理论研究,通过对Bulk体系以及Device体系电子结构以及输运性质分析,得出以下结论:
(1) 当对交错构型的MoS2纳米带进行苯分子插层功能化时,纳米带材料会由原来的半导体性转变为金属性,在0 eV附近出现跨越费米能级的能带。
(2) 由Kohn-Sham轨道和电子差分密度图可知,由于苯环和MoS2纳米带之间的相互作用,苯分子参与到电子输运过程中,增加了输运过程中载流子的浓度。
(3)I-V特性曲线和透射光谱表明,苯分子功能化之后的MoS2纳米带的导电性得到了增强,从大到小依次为MoS2@βC6H6> MoS2@αC6H6> MoS2,这与偏压窗口中透射峰面积是对应的。
综上所述,通过将有机分子插入MoS2纳米带层间对其功能化以提高其输运性质是可行的,这也为未来的理论和实验工作提供了参考。
猜你喜欢
中学化学(2022年5期)2022-06-17
建筑材料学报(2021年2期)2021-05-15
高中数理化(2020年1期)2020-02-29
四川师范大学学报(自然科学版)(2018年2期)2018-04-28
材料科学与工程学报(2016年2期)2017-01-15
通信电源技术(2016年6期)2016-04-20
中国塑料(2015年6期)2015-11-13
无机化学学报(2014年12期)2014-02-28
河南科技(2014年11期)2014-02-27
东北石油大学学报(2014年3期)2014-02-27
