
液相色谱串联质谱法测定保健食品中3种甲基咪唑类物质
2021-07-06 11:11王德利冷佳蔚胡文红
化工设计通讯 2021年6期
王德利,冷佳蔚,姜 俊,胡文红
(1.山东省食品药品检验研究院,山东济南 250101;2.山东省食品药品安全检测工程技术研究中心,山东济南 250101)
焦糖色素是食品生产中常用的一种黑色着色剂,颜色呈黑褐色,经常用于加入酱油和可乐中的一种用于改变颜色的添加剂[1],其实焦糖色素也经常被用于以中草药提取物为原料生产的保健食品剂型中。焦糖色素生产时会产生致癌小分子副产物4-甲基咪唑(4-MI)[2]。根据世界卫生组织研究发现,4-MI在动物生长过程中能诱发动物肿瘤的生长,同时此类小分子化合物也有可能致癌。1-甲基咪唑与2-MI和4-MI互为同分异构体。
保健食品成分复杂含有多种添加剂、植物源性提取成分等,易造成定量不准确,常用的样品前处理方法有QuEchers法[3]和固相萃取法[4]等。液相色谱方法定量检测时,基质复杂定性不准确,同时灵敏度不能达到痕量分析的要求,气相色谱方法在保健食品前处理过程中目标化合物需要进行衍生化,实验过程复杂耗时较长,光谱方法和电化学方法抗干扰能力差,定性和定量不准确,因此不适合选择这些方法。目前,对于保健食品中的1-MI、2-MI和4-MI则没有液相色谱串联质谱同时检测的方法和数据。
建立一种液相色谱质谱法测定保健食品中的甲基咪唑类化合物,通过固相萃取净化方式对保健食品进行处理,3种甲基咪唑类化合物同时进行定量,并结合亲水色谱保留模式,对保健食品中3种甲基咪唑类化合物进行分离,串联质谱法能够进行定量。
1 材料与方法
1.1 材料与试剂
1-甲基咪唑和4-甲基咪唑标准物质:北京曼哈格生物科技有限公司;2-甲基咪唑标准物质:上海安谱实验科技股份有限公司;1-甲基咪唑-氘代内标,2-甲基咪唑-氘代内标,4-甲基咪唑-氘代内标标准物质:加拿大C/D/N ISOTopes 公司;甲醇,乙腈,甲酸铵:色谱纯,德国Merck公司。
1.2 仪器与设备
H-Class超高效液相色谱仪:美国waters公司;SCIEX 5500质谱仪:美国AB SCIEX公司;旋涡混合器:德国艾卡公司;离心机:德国西格玛公司;氮吹仪:美国Organomation公司。
1.3 标准溶液配制
储备液配制:称取3种标准物质10mg,用乙腈溶解定容至10mL,配制成1mg/mL的标准储备液。
内标储备液配制:称取3种内标标准物质10mg,用乙腈溶解并定容至10mL,配制成1mg/mL的内标标准储备液。
配制标准工作曲线:移取适量标准溶液和1μg/mL同位素内标溶液,配制成10、50、100、200、500ng/mL系列的含50ng/mL内标溶液的标准工作溶液。
1.4 处理方法
称取2.0g样品,加100μL 1μg/mL内标液,加入1%三氯乙酸20mL混合均匀,进行超声提取30min,在离心机8 000r/min高速离心5min后,把离心后的上清液取出放入另一离心管中。准确移取10mL过阳离子固相萃取柱,分别用5mL 2%甲酸淋洗和5mL甲醇各淋洗一次,用10mL 5%氨水甲醇洗脱,收集洗脱液,45 ℃氮吹,加1.0mL初始流动相复溶,过膜,上机测定。
1.4.1 色谱条件
色谱柱:BEH HILIC;柱温:40 ℃;流速:0.3mL/min;体积:2μL;流动相:A为10mmol/L甲酸铵,B为乙腈。梯度程序如表1所示。

表1 流动相梯度程序
1.4.2 质谱条件
质谱离子源:ESI+;检测方式:MRM监测;气帘气:20.0L/h;碰撞气:8L/h;离子喷雾电压:5kV;离子源温度:500 ℃;喷雾气流速:50L/h;辅助气流速:50L/h。
2 结果与分析
2.1 固相萃取柱的选择
保健食品样品基质比较复杂,为了提高检测的灵敏度和准确度,选择使用固相萃取柱进行样品除去杂质和富集目标物。选择不同保留机理的4种固相萃取柱C18、MCX、HLB和MAX对3种甲基咪唑类化合物进行回收率试验,如图1所示。结果表明在HLB和MAX固相萃取柱上回收率在20%以下,几乎没有保留,由于3种甲基咪唑类化合物在酸性溶液中成为阳离子状态,在HLB和MAX阴离子固相萃取柱上无法保留,导致回收率较低。3种化合物在C18固相萃取柱上回收率不超过60%,说明目标物在C18固相萃取柱上保留能力也比较差,损失严重,最终导致定量结果偏差较大。当使用MCX固相萃取柱时回收率能达到90%以上,说明3种甲基咪唑类化合物在酸性溶液中以阳离子存在,因此在MCX阳离子固相萃取柱中目标物能够以离子作用力保留,因此在进一步的样品前处理过程中选择使用MCX固相萃取柱。

图1 3种甲基咪唑在固相萃取柱中的回收率
2.2 固相萃取柱洗脱条件的优化
考察洗脱溶液甲醇中氨水体积分数对3种甲基咪唑化合物的洗脱能力,结果如图2所示。从图2可以看出,氨水比例5%时回收率最高,但当氨水比例继续增大时,回收率反而有所降低。分析可能是因为当洗脱溶液为碱性时,甲基咪唑类化合物以分子形式存在,能够从MCX阳离子固相萃取柱上洗脱下来。通过以上分析可以得出选择体积分数5%氨水作为洗脱溶液最合适。

图2 洗脱液中氨水比例优化
2.3 色谱分离条件的优化
通过化合物结构分析,3种甲基咪唑类化合物极性较强,适合使用亲水性的BEH HILIC色谱柱进行分离,在实际分离3种目标化合物时分离度和色谱峰性都很好,另3种目标物在一种亲水的BEH Amide色谱柱上色谱峰有明显拖尾和色谱峰展宽现象,导致在定量时造成结果偏差较大。亲水模式色谱与质谱串联兼容性更好,对3种甲基咪唑类化合物同时分离效果会更好,灵敏度也将得到极大提高,因此选择BEH HILIC亲水色谱柱进行分离检测。
2.4 质谱条件的优化
3种甲基咪唑产生m/z 56.1和m/z 42.1的碎片离子。碎片离子分别选择作为定性与定量离子对,并通过优化质谱参数,使碎片离子响应强度达到最大。经优化后,3种甲基咪唑化合物及同位素内标物关键质谱参数如表2所示。
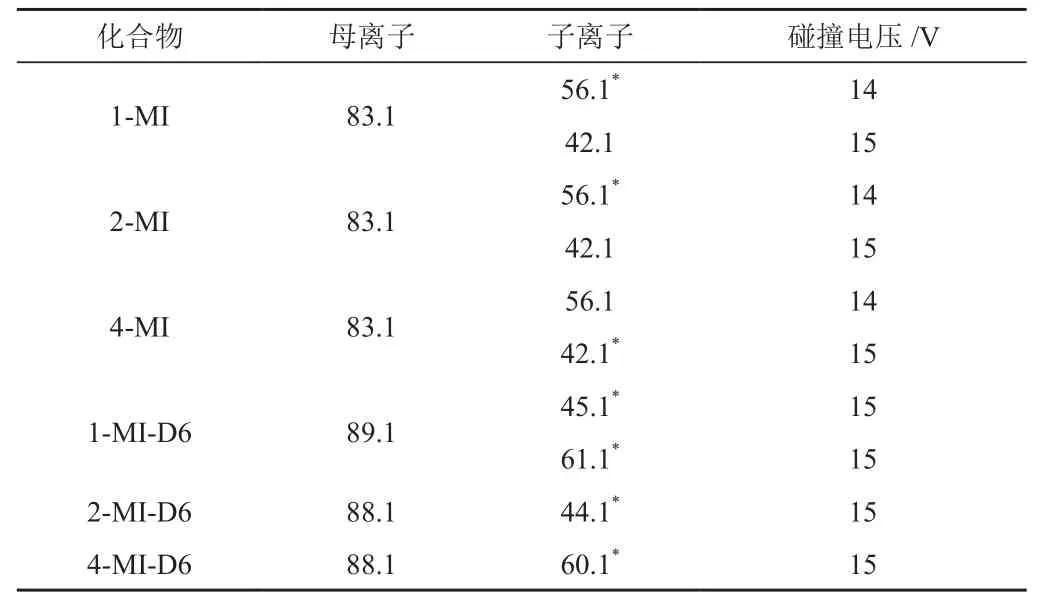
表2 3种甲基咪唑及同位素内标质谱参数
2.5 方法的线性范围和检出限
以化合物定量离子与内标定量离子峰面积比值为纵坐标,以标准溶液浓度(ng/mL)为横坐标,绘制标准曲线,结果,如表3所示。

表3 3种甲基咪唑线性方程、线性相关系数和检出限
2.6 方法的回收率和精密度
采用两种不含3种甲基咪唑类化合物的不同类型保健食品,进行添加回收率和精密度的试验。保健食品样品中添加10、50和100μg/kg三浓度水平,计算回收率和相对标准偏差,3种甲基咪唑化合物在不同基质的回收率在95%~106.2%,RSD在2.1%~5.5%。
2.7 实际样品测定
用本文建立的方法对市场上购买的50批次保健食品样品进行检测。其中保健食品中1-MI未检出,20批次2-MI含量最高为303.7μg/kg,25批次4-MI含量最高为3 510.4μg/kg。通过对比配料表总结出,配料中含有焦糖色素的保健食品有26批次,其中4-MI检出有25批次,2-MI检出有20批次。通过检测结果可以看出含有焦糖色素的检出率很高,可能是因为在焦糖色素生产过程中产生了焦糖化,生成了有害的副产物甲基咪唑类化合物,可能不同工艺生产的焦糖色素产生的副产物含量也不同,因此保健食品中甲基咪唑类化合物的危害性有待评估。
3 结束语
通过对实验条件进行优化,最终选择提取溶液为浓度1%的三氯乙酸,净化固相萃取柱为MCX,该提取净化方法能够提高方法的灵敏度和准确性,选择BEH HILIC亲水模式色谱柱,建立了液相色谱串联质谱法测定保健食品中1-MI、2-MI和4-MI内标定量分析方法。
猜你喜欢
绿色科技(2021年12期)2021-07-22
化工自动化及仪表(2021年3期)2021-06-04
食品工程(2020年4期)2021-01-20
中华养生保健(2020年3期)2020-11-16
农药科学与管理(2019年9期)2019-11-23
酿酒科技(2019年10期)2019-11-12
科技视界(2016年26期)2016-12-17
发明与创新(2016年14期)2016-06-09
发明与创新·中学生(2016年4期)2016-05-14
妇女生活(2016年4期)2016-05-03
