
lncRNA SNHG6调控miR-335-3p/PDCD4途径对缺氧复氧诱导的心肌细胞损伤的影响
2021-08-03 12:22尤其郭辉
心电与循环 2021年4期
尤其 郭辉
溶栓、经皮冠状动脉介入治疗是心肌梗死后恢复血供的有效方法,但随着血供恢复会造成额外的心肌损伤,即心肌缺血/再灌注(ischemia/reperfusion,I/R)损伤。因此,减轻I/R损伤对改善心肌梗死患者预后具有重要意义。近年来,长链非编码RNA(long non-coding RNA,lncRNA)受到人们的关注,其通过与靶微小RNA(microRNA,miRNA)结合抑制miRNA对靶mRNA表达的负调控,具有多种生理和病理功能[1]。目前,已有多种lncRNA参与心肌I/R损伤的调控[2-3]。研究报道,在缺血性脑卒中体内外模型中lncRNA小核RNA宿主基因6(small nuclear RNA host gene 6,SNHG6)表达增加,抑制SNHG6表达可提高神经元细胞活力,抑制氧糖剥夺诱导的细胞凋亡,减少脑梗死面积,并改善神经系功能[4]。然而,SNHG6在心肌I/R损伤中的作用并不清楚。DIANA工具预测到微小RNA-335-3p(microRNA-335-3p,miR-335-3p)与SNHG6存在特异结合位点。已有研究表明,在小鼠局灶性脑I/R模型中,敲除环状RNA丝氨酸/苏氨酸蛋白扰动样激酶1(circular RNATousled-like Kinase1,circTLK1)通过上调miR-335-3p表达减少梗死体积,可减轻神经元损伤,改善神经功能缺损[5]。Targetscan预测到程序性细胞凋亡因子4(programmed cell death 4,PDCD4)与miR-335-3p存在特异结合位点,在I/R心肌细胞中PDCD4表达上调,下调PDCD4可增强心肌细胞活力,促进细胞周期进程,抑制细胞凋亡[6]。但SNHG6/miR-335-3p/PDCD4分子轴在心肌I/R损伤中的作用尚未阐明。本研究旨在研究SNHG6在心肌I/R损伤中的作用,并通过miR-335-3p/PDCD4进一步探讨其作用机制。
1 材料和方法
1.1 细胞、试剂和仪器 大鼠心肌细胞系H9C2购自中国武汉典型培养物保藏中心;杜尔伯格氏伊戈尔培养基(dulbecco's modified eagle medium,DMEM)及胎牛血清购自美国Gibco公司;SNHG6小干扰RNA(si-SNHG6)、miR-335-3p模 拟 物(mimics)、miR-335-3p抑制物(anti-miR-335-3p)以及相应阴性对照(si-NC、miR-NC、anti-miR-NC)购自北京六合华大基因公司;miRNA提取试剂盒(200116)购于北京天根生化科技公司;M-MLV逆转录酶(200101)购自美国Life Technologies公司;SYBR Green PCR Master Mix(191028)购自美国ABI公司;miRNA反转录试剂盒(190812)、miRNA检测试剂盒(191201)、膜联蛋白V-异硫氰酸荧光素(Annexin-V-FITC)/碘化丙啶(propidium iodide,PI)凋亡检测试剂盒(191011)购自北京百奥莱博生物公司;小鼠PDCD4单克隆抗体(sc-376430)、小鼠磷酸甘油醛脱氢酶(glyceraldehyde phosphate dehydrogenase,GAPDH)单克隆抗体(sc-47724)、羊抗鼠IgG二抗(sc-2005)购自美国Santa Cruz公司;鼠源裂解的半胱氨酸蛋白酶3(Cleaved Caspase-3)多克隆抗体(ab2302)、羊抗兔IgG二抗(ab97051)购于美国Abcam公司;超氧化物歧化酶(superoxide dismutase,SOD)活性检测试剂盒(190911)、丙二醛(malondialdehyde,MDA)检测试剂盒(191005)购自北京索莱宝生物公司;双荧光素酶活性测定试剂盒(200201)购自北京天恩泽基因科技公司。荧光定量PCR仪(7500型)购自美国ABI公司;流式细胞仪(FACSCalibur)购自美国BD公司。
1.2 方法
1.2.1 细胞培养、转染和分组 细胞培养:参照李瑞萍等[7]实验方法建立心肌细胞缺氧复氧损伤模型,H9C2细胞接种于无血清低糖(糖浓度1.0 g/L)DMEM培养液,放入缺氧培养箱(95%N2,5%CO2)培养3 h,然后更换为含10%胎牛血清的DMEM高糖(糖浓度4.5 g/L)培养液,放入正常培养箱(95%空气,5%CO2)复氧培养6 h。细胞转染:将对数期H9C2细胞以2×105个/孔接种于6孔板。利用脂质体Lipofectamine 2000将si-SNHG6、si-NC、miR-335-3p mimics、miR-NC、si-SNHG6+anti-miR-NC、si-SNHG6+anti-miR-335-3p分别转染汇合度为60%的H9C2细胞,收获转染48 h细胞。分组:对照组为正常培养的H9C2细胞;模型组为缺氧处理3 h、复氧处理6 h的H9C2细胞;模型+si-NC组、模型+si-SNHG6组、模型+miR-NC组、模型+miR-335-5p组分别为转染si-NC、转染si-SNHG6、转染miR-NC、转染miR-335-5p mimics的H9C2细胞(缺氧处理3 h、复氧处理6 h);模型+si-SNHG6+anti-miR-NC组、模型+si-SNHG6+anti-miR-335-5p组分别为转染si-SNHG6与anti-miR-NC、转染si-SNHG6与anti-miR-335-5p的H9C2细胞(缺氧处理3 h、复氧处理6 h)。
1.2.2 实时定量PCR(real-time quantitative PCR,RT-qPCR)检测SNHG6、miR-335-3p表达TRIzol试剂盒、miRNA提取试剂盒分离H9C2细胞的总RNA和miRNAs。M-MLV逆转录酶合成cDNA,再用SYBR Green PCR Master Mix进行荧光定量PCR以检测SNHG6表达水平。采用miRNA反转录试剂盒合成互补DNA(complementary DNA,cDNA),采用miRNA检测试剂盒进行荧光定量PCR以检测miR-335-3p表达水平。SNHG6、miR-335-3p分别以GAPDH和U6作 为 内 参 对 照,2-ΔΔCt法 计 算SNHG6、miR-335-3p表达量。引物序列如下(5'-3'):SNHG6上游ATACTTCTGCTTCGTTACCT,下游CTCATTTTCATCATTTGCT;GAPDH上游GGGAGCCAAAAGGGTCATCA,下游TGATGGCATGGACTGTGGTC;miR-335-3p上游UUUUUCAUUAUUGCUCCUGACC,下游CCAGTCTCAGGGTCCGAGGTATTC;U6上游CTCGCTTCGGCAGCACA,下游AACGCTTCACGAATTTGCGT。
1.2.3 流式细胞术检测细胞凋亡H9C2细胞处理完毕后,收集细胞,PBS洗涤细胞2次,并用结合缓冲液重悬细胞。取100 μL细胞悬液(细胞数约1×105个),加入Annexin-V-FITC和PI各5 μL,暗室内孵育30 min后添加400 μL结合缓冲液,混匀后上流式细胞仪检测凋亡细胞百分比。
1.2.4 分光光度法检测细胞中SOD活性、MDA水平 缺氧复氧处理结束后,收集细胞至离心管,加入提取液超声破碎细胞,收集上清液,按照商品试剂盒说明书测定SOD活性、MDA水平。
1.2.5 蛋白质印记法检测PDCD4和Cleaved Caspase-3蛋白表达 放射免疫沉淀试验(radioimmunoprecipitation assay,RIPA)裂解液提取H9C2细胞总蛋白。取等量的蛋白样品进行十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE),然后转移蛋白至聚偏二氟乙烯膜上,封闭膜后与特异性一抗(PDCD4和GADPH均 为1∶300稀 释,Cleaved Caspase-3为1∶500稀释)在4℃孵育过夜。室温下将膜与对应二抗孵育1 h。将膜置于孵育盒,滴加化学发光试剂暗室显影。Image-Pro Plus 6.0软件分析PDCD4蛋白条带和GADPH条带灰度值,二者的比值表示PDCD4蛋白表达量。
1.2.6 双荧光素酶报告实验 将含有miR-335-3p结合位点的野生型(wild type,WT)或突变型(mutant type,MUT)SNHG6或PDCD4-3'-UTR序列克隆至psiCHECK-2载体,构建荧光素酶报告质粒WTSNHG6、MUT-SNHG6、WT-PDCD4、MUT-PDCD4。利用脂质体转染法将上述报告质粒分别与miR-335-3p mimics(miR-335-3p组)、miR-NC(miR-NC组)转染H9C2细胞,双荧光素酶活性测定试剂盒分析转染48 h后各组细胞相对荧光素酶活性。
1.3 统计学处理 采用SPSS 18.0统计软件,正态分布的计量资料以表示,多组比较采用单因素方差分析,组间两两比较采用LSD-t检验。P<0.05为差异有统计学意义。
2 结果
2.1 各组细胞SNHG6、miR-335-5p、PDCD4、Cleaved Caspase-3表达水平及凋亡等指标的比较 见图1、表1。
由图1、表1可见,与对照组比较,模型组SNHG6表达量,PDCD4、Cleaved Caspase-3蛋白表达量,细胞凋亡率及MDA水平均显著升高(均P<0.01),miR-335-5p表达量、SOD活性均显著降低(均P<0.01);与模型+si-NC组比较,模型+si-SNHG6组SNHG6表达量,PDCD4、Cleaved Caspase-3蛋白表达量,细胞凋亡率及MDA水平均显著降低(均P<0.01),miR-335-5p表达量、SOD活性均显著升高(均P<0.01)。
表1 各组细胞SNHG6、miR-335-5p、PDCD4、Cleaved Caspase-3表达水平及凋亡等指标的比较

图1 干扰SNHG6对细胞凋亡流式图及PDCD4蛋白表达电泳图的影响
2.2 双荧光素酶报告实验结果DIANA工具预测到miR-335-5p与SNHG6序列存在连续特异性结合位点,Targetscan预测到miR-335-5p与PDCD4-3'-UTR区域存在连续特异性结合位点,见图2。双荧光素酶报告实验显示,与miR-NC组比较,miR-335-5p组WT-SNHG6、WT-PDCD4的相对荧光素酶活性均显著下降(均P<0.01),而MUT-SNHG6、MUT-PDCD4的相对荧光素酶活性差异均无统计学意义(均P>0.05),见表2。
表2 双荧光素酶报告实验结果

图2 小核RNA宿主基因6(SNHG6)、微小RNA(miR)-335-5p和程序性细胞凋亡因子4(PDCD4)的互补序列
2.3 各组细胞miR-335-5p、PDCD4、Cleaved Caspase-3蛋白表达水平及凋亡等指标的比较 见图3、表3。
由图3、表3可见,与模型+miR-NC组比较,模型+miR-335-5p组miR-335-5p表达量、SOD活性均显著升高(均P<0.01),PDCD4和Cleaved Caspase-3蛋白表达量、细胞凋亡率、MDA水平均显著降低(均P<0.01)。
表3 各组细胞miR-335-5p、PDCD4、Cleaved Caspase-3蛋白表达水平及凋亡等指标的比较
图3 微小RNA(miR)-335-5p对细胞凋亡流式图及程序性细胞凋亡因子4(PDCD4)蛋白表达电泳图的影响
2.4 各 组 细 胞miR-335-5p、PDCD4、CleavedCaspase-3表达及凋亡等指标的比较 见图4、表4。
由图4、表4可见,与模型+si-SNHG6+antimiR-NC组比较,模型+si-SNHG6+anti-miR-335-5p组miR-335-5p表达量、SOD活性均显著降低(均P<0.01),PDCD4和Cleaved Caspase-3蛋白表达水平及细胞凋亡率、MDA水平均显著升高(均P<0.01)。
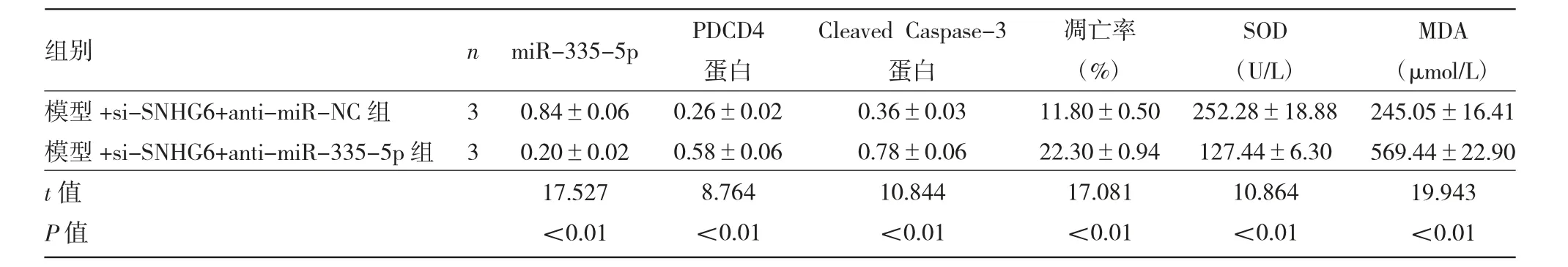
表4 各组细胞miR-335-5p、PDCD4、Cleaved Caspase-3表达及凋亡等指标的比较

图4 抑制微小RNA(miR)-335-5p可逆转干扰小核宿主基因6(SNHG6)对凋亡流式图及程序性细胞凋亡因子4(PDCD4)蛋白表达电泳图的影响
3 讨论
lncRNA通过调控基因表达参与心肌I/R损伤的多种病理过程,具有作为心肌I/R损伤治疗靶点的潜力。Du等[8]研究指出lncRNA核富集转录体1(NEAT1)/miRNA-520a轴参与心肌I/R损伤中线粒体途径细胞凋亡的调控。Xue等[9]的研究表明,lncRNA低氧诱导因子1α反义RNA1(HIF1AAS1)通过吸附miRNA-204调节信号转导蛋白抑制因子2表达参与心肌I/R损伤后心室重构。Liang等[10]报道抑制lncRNA ROR可减弱缺氧复氧诱导的心肌细胞凋亡和炎症反应。然而,SNHG6在心肌I/R损伤中功能并未阐明。已有文献证实SNHG6在结直肠癌、乳腺癌等实体肿瘤中表达上调,具有致癌因子作用[11-12]。SNHG6表达增加还可能与室间隔缺损形成有关[13]。本研究证实缺氧复氧处理后H9C2细胞中SNHG6表达量明显增加,提示SNHG6表达改变可能与心肌I/R损伤有关。功能实验表明,转染si-SNHG6干扰SNHG6表达显著抑制缺氧复氧诱导的细胞凋亡,抑制Cleaved Caspase-3蛋白表达。心肌缺氧后,尤其在复氧阶段产生大量的活性氧,SOD是清除活性氧的重要氧抗氧化酶,当机体氧化抗氧化失衡时导致活性氧攻击生物膜诱导广泛的氧化应激损伤[14-15]。本研究中干扰SNHG6表达后,缺氧复氧诱导H9C2细胞中SOD活力明显增加,脂质过氧化终产物MDA水平明显降低,说明干扰SNHG6表达能够减弱缺氧复氧诱导的心肌细胞氧化应激损伤。Shan等[16]证实在动脉粥样硬化中,敲减SNHG6能够提高人脐静脉内皮细胞活力,减弱低密度脂蛋白诱导的氧化应激、炎症和细胞凋亡,这与本研究中干扰SNHG6表达的保护作用一致。以上数据表明,SNHG6可能参与心肌I/R损伤。
研究表明lncRNA通过表观遗传、转录或翻译机制参与人类心血管疾病进展[17]。对SNHG6进行分析发现其功能的发挥多与调控miRNA表达有关,例如SNHG6通过靶向miR-101-3p参与胶质瘤细胞的增殖、上皮-间充质转化和迁移,抑制细胞凋亡[18]。SNHG6靶向miR-26a-5p调控结直肠癌细胞的5-氟尿嘧啶耐药性[19]。本研究通过荧光素酶实验证实SNHG6与miR-335-3p直接存在相互作用,进一步研究显示缺氧复氧处理后H9C2细胞中miR-335-3p表达明显降低,而干扰SNHG6表达显著减弱缺氧复氧对miR-335-3p表达的抑制作用,提示SNHG6可能靶向miR-335-3p参与心肌I/R损伤。转染miR-335-3p mimics进行功能实验,结果表明过表达miR-335-3p显著减轻缺氧复氧诱导的H9C2细胞凋亡和SOD活力下降,下调Cleaved Caspase-3蛋白表达并降低MDA水平,这与Wu等[5]报道的miR-335-3p在脑I/R损伤中的保护作用一致。miRNA主要通过识别靶mRNA抑制其翻译来调控蛋白表达进而发挥作用。PDCD4是细胞凋亡的关键介质,正常心肌组织中PDCD4表达水平较低,过氧化氢或缺氧复氧处理后心肌细胞中PDCD4表达上调可加重细胞凋亡,通过下调PDCD4表达可改善过氧化氢诱导的心肌细胞损伤和心肌I/R损伤[20-22]。本研究发现缺氧复氧处理后H9C2细胞中PDCD4表达增加,并证实PDCD4是miR-335-3p的靶基因,且干扰SNHG6或过表达均可减弱缺氧复氧处理对PDCD4表达的促进作用,这说明SNHG6/miR-335-3p/PDCD4途径可能在心肌I/R损伤中发挥作用。回复实验显示,抑制miR-335-3p表达显著减弱干扰SNHG6表达对H9C2细胞缺氧复氧损伤的影响,这说明SNHG6通过靶向miR-335-3p进而上调PDCD4表达参与缺氧复氧诱导的心肌细胞损伤。
总之,本研究证实干扰SNHG6表达通过上调miR-335-3p/PDCD4途径能够减弱缺氧复氧诱导的心肌细胞凋亡和氧化应激损伤,这进一步揭示了心肌I/R损伤中lncRNA/miRNA/mRNA调控机制,为心肌I/R损伤提供潜在治疗靶点。
猜你喜欢
传染病信息(2022年3期)2022-07-15
法医学杂志(2022年1期)2022-06-21
中西医结合心脑血管病杂志(2022年4期)2022-03-11
中西医结合心脑血管病杂志(2022年2期)2022-02-15
保健与生活(2020年6期)2020-03-20
江苏农业科学(2019年23期)2019-03-03
体育科学(2018年12期)2019-01-04
中国卫生产业(2018年12期)2018-05-14
中国中医药信息杂志(2018年11期)2018-01-05
三联生活周刊(2017年1期)2017-01-11
