
苦荞WOX家族全基因组鉴定及响应愈伤诱导率表达分析
2021-09-18 06:16侯思宇王欣芳杜伟冯晋华韩渊怀李红英刘龙龙孙朝霞
中国农业科学 2021年17期
侯思宇,王欣芳,杜伟,冯晋华,韩渊怀,李红英,刘龙龙,孙朝霞
苦荞WOX家族全基因组鉴定及响应愈伤诱导率表达分析
侯思宇1,王欣芳1,杜伟1,冯晋华1,韩渊怀1,李红英1,刘龙龙2,孙朝霞1
1山西农业大学农学院,山西太谷 030801;2山西农业大学农业基因资源研究中心,太原 030031
【】全基因组鉴定苦荞WOX(WUSCHEL-related homeobox)基因,揭示其基因家族成员序列特征、基因表达模式及与出愈率的相关性,为突破苦荞再生及遗传转化难题提供理论基础。基于同源性搜索策略,以拟南芥WOX基因蛋白为参考序列,进行苦荞全基因组比对,获得苦荞WOX基因家族成员蛋白及核酸序列。基于蛋白同源性及保守结构域分析,鉴定出苦荞WOX基因家族所有成员。同时使用TBtools软件展示FtWOXs家族成员基因结构、保守结构域及启动子顺式作用元件特征。比较分析WOX基因家族成员在苦荞与拟南芥之间的基因组共线性。基于邻近法,利用MEGA X软件构建苦荞、拟南芥和水稻WOX基因家族成员蛋白序列系统进化树。以MS+2,4-D 3.0 mg·L-1+6-BA 1.0 mg·L-1为愈伤诱导培养基,下胚轴为外植体,选取70份苦荞品种诱导愈伤组织,评价不同基因型的出愈率。qRT-PCR比较分析高、低出愈率苦荞品种间FtWOXs基因表达水平。基于Pearson相关系数分析出愈率与FtWOXs基因家族成员表达相关性。共鉴定出30个苦荞WOX基因成员,在苦荞8条染色体上呈现不均匀分布。系统进化树表明30个苦荞WOX基因可划分为3大类,不同类群中WOX基因包含不同的保守结构域,主要的保守结构域为HD(Homeodomain)、START和MEKHLA结构域。保守基序分析表明,FtWOXs基因家族成员所含保守基序数目的范围为2—10个。基因结构分析表明,FtWOXs基因家族成员所含外显子数目的范围为2—18个。顺式作用元件分析表明FtWOXs基因启动子富含26个不同种类的顺式作用元件。系统进化分析表明,30个苦荞、15个拟南芥和12个水稻WOX基因家族成员可分为3类,其中第3类为苦荞独有。基因组共线性分析表明,6个WOX基因在苦荞和拟南芥之间存在基因组共线性。表达模式及相关性表明,////与苦荞出愈率存在正相关性。苦荞FtWOXs成员存在丰富的序列变异特征,不同苦荞基因型中WOX基因表达水平及出愈率存在明显差异和一定的相关性,揭示不同苦荞WOX基因具有潜在的功能多样性。
苦荞;WOX基因;愈伤组织;基因表达
0 引言
【研究意义】荞麦为蓼科荞麦属,有普通荞麦(甜荞)和鞑靼荞麦(苦荞)2个栽培种,广泛种植于俄罗斯、中国、哈萨克斯坦、日本、法国等国家,栽培历史悠久[1]。荞麦生长周期短、耐瘠薄、用途广,是适合复式种植的可持续发展作物;其营养价值丰富,富含生物活性化合物,如荞麦糖醇(D-手性肌醇)、黄酮类(主要是芦丁、槲皮素)、多酚等,具有降糖、抗癌、消炎、降低胆固醇及高血压、心脏病发病率等作用[2]。由于苦荞为自花授粉作物,很多优良性状基因无法通过杂交方式转入,一定程度上限制了苦荞育种的进程,造成苦荞优良品种不足[3]。随着育种水平的发展,利用基因工程手段进行种质创新成为研究热点,高效离体再生体系的建立是其中重要的一环。【前人研究进展】植物离体再生的过程实质上是决定细胞命运的关键基因被激活或关闭的过程,涉及基因包括()、()、()和2等[4]。其中,WUSCHEL(WUS)-related homeobox(WOX)转录因子家族典型序列特征为包含Homeobox(HB)蛋白(WUS基因是该家族中最早发现的成员),其同源异型结构域(homeodomain,HD)由65个氨基酸残基组成,属于锌指蛋白(ZIP)超家族。早在1994年,HD蛋白就被证实是真核生物生长发育调节的关键因子[5-6]。在全面研究拟南芥基因组后,发现了15个与WUS基因结构相似的基因,并将它们命名为WOX基因,这些基因是植物干细胞维持、胚胎建成和器官发生等发育过程中的重要调控因子[7-8]。在细胞和组织培养研究的芽再生过程中,证明WUS基因的表达是干细胞分化所必需的[9-10]。拟南芥WUS基因能够促进体细胞向胚性细胞的转变,其功能获得性突变体的外植体能够在不依赖于外源植物激素的条件下,形成大量体细胞胚[11]。在葡萄体细胞胚再生过程中,和在鱼雷胚和子叶发生阶段被激活,调控体细胞发生[12]。同时WOX基因具有多种功能,如维持根尖分生组织活性[13],是早期胚和子叶形成的必需因子[14]。研究表明,WOX基因家族在植物进化及应对环境胁迫方面都起到了重要作用,且各成员间分工不同。如是重要的冷胁迫响应因子[15];在水稻中,WOX基因也参与调节干旱、盐和冷胁迫[16]。【本研究切入点】苦荞次生代谢物质较多,在组织培养过程中易褐化、形成严重的不定根,且对基因型的依赖性很强,不同品种之间愈伤组织的诱导能力有明显差异。WOX基因已被证实在拟南芥、水稻等植物的遗传再生方面具有重要作用,但在苦荞中的作用仍鲜见报道。【拟解决的关键问题】本研究采用生物信息学分析方法,全基因组鉴定苦荞WOX基因家族成员,拟寻找可能导致苦荞再生能力差异的基因;在此基础上对不同基因型苦荞WOX基因表达模式与出愈率之间的相关性进行分析,以期筛选到影响愈伤组织增殖的关键基因,为苦荞高效再生体系的建立及遗传转化研究提供技术支持。
1 材料与方法
1.1 苦荞WOX基因家族成员核酸和蛋白序列查找及理化性质分析
从Tair网站在线获取拟南芥的15个WOX基因成员蛋白序列,并下载至本地。从Pfam数据库中搜索Pfam ID号(PF00046)及隐马尔可夫模型文件。以拟南芥WOX基因蛋白序列为种子序列,blast苦荞基因组数据库注释的蛋白序列,将蛋白同源性高于1×e-5的苦荞WOX基因序列下载至本地用于进一步分析,每个WOX基因成员如果有多个高同源性的基因,只选择同源性Top1的序列。为保证获得全基因组范围内WOX基因家族所有成员,将上述获得的苦荞WOX基因家族成员蛋白序列反向翻译为核酸序列,进一步比对苦荞全基因组核酸序列,以免漏掉未注释的WOX基因。进一步通过保守结构域数据库比对分析(CDD:https://www.ncbi.nlm.nih.gov/Structure/ cdd/wrpsb.cgi)[17],过滤结构域不完整以及氨基酸数目小于100的基因,最终鉴定出30个苦荞WOX基因。以上结果采用TBtools[18]软件进行批处理分析。利用ExPASy网站(https://web.expasy.org/protparam/)对WOX蛋白序列的氨基酸数目、等电点、稳定性及分子量等进行分析;使用PredictProtein(https:// predictprotein.org/)、ProtComp9.0(http://linux1. softberry.com/berry.phtml?topic=protcomppl&group=programs&subgroup=proloc)和SignalIP5.0(http://www. cbs.dtu.dk/services/SignalP/)对WOX基因进行二级结构预测、亚细胞定位和信号肽预测。同时采用同源性比对分析,从NCBI(https://www.ncbi.nlm.nih.gov/)数据库搜索并获得水稻WOX基因家族成员序列信息和Genbank ID,最终获得12个水稻WOX基因。
1.2 系统发育树构建及保守结构域分析
对15个拟南芥、12个水稻和30个苦荞WOX基因编码蛋白序列,利用Clustal X[19]软件进行多重序列比对分析,序列比对后的矩阵用于系统进化树分析,利MEGA X[20]软件邻近法(Neighbor-Joining,NJ)构建系统进化树(设置参数Bootstrap检验值为10 000)。在Batch sequence search(http://pfam.xfam.org/search# tabview =tab1)在线结构域分析网站,提交30个苦荞WOX基因编码蛋白序列,获得保守结构域特征,并利用TBtools软件可视化保守结构域信息。
利用MEME(http://meme-suite.org)在线预测分析苦荞WOX基因编码蛋白的保守基序[21]。基于PLANTCARE[22]数据库(http://bioinformatics.psb.gent. e/eboolslantcare/html)在线分析启动子元件。使用TBtools子程序gene structure viewer可视化分析苦荞WOX基因家族所有成员的基因结构、保守基序和顺式作用元件。
1.3 苦荞WOX基因家族基因组共线性分析及选择压力分析
使用TBtools子程序BLAST GUI Wrapper对苦荞和拟南芥WOX基因共线性结果进行可视化,同时进化选择压力分析,获得共线性基因的非同义替换率(Ka)、同义替换率(Ks)和Ka/Ks比值。
1.4 愈伤组织的诱导及继代培养
试验材料为团队前期收集到不同地理来源的苦荞种质资源70份(表1)。外植体培养及愈伤组织诱导步骤如下:将籽粒饱满的种子置于0.1% Tween 20溶液中浸泡20 min;随后加入15 mL 5%次氯酸钠溶液振荡5 min,重复2次;用灭菌水冲洗3—5次,置于无菌滤纸晾干后接种于MS培养基中,每瓶接种30粒,每个品种重复3次,于(25±2)℃条件下暗培养。待下胚轴伸长至3.0—5.0 cm时,切取0.5—1.0 cm小段作为外植体用于愈伤组织培养。将外植体接种于MS+3.0 mg·L-12,4-D+1.0 mg·L-16-BA培养基中,设置3组重复(每组接种25个下胚轴);暗培养7 d后转至光下,光周期为16 h光/8 h暗,光照强度为37.5 μmol·m-2·s-1,相对湿度保持在60%—80%,温度(25±2)℃。培养过程中观察出愈变化、愈伤组织颜色、形态及质地,外植体接种3周后统计愈伤组织和胚性愈伤组织的个数,以下胚轴膨大约2 cm为愈伤组织诱导成功标准,计算出愈率。筛选出高(>95%)和低(<30%)出愈率品种的愈伤组织为试验材料,用于RNA提取及基因表达模式分析。
出愈率(%)=诱导出愈伤组织的外植体数/外植体总数×100。

表1 用于愈伤组织诱导的苦荞品种
1.5 样品总RNA提取
使用总RNA提取试剂盒(TIANGEN,北京,DP419)提取样品RNA,琼脂糖凝胶电泳检测完整性及NanoDrop2000(Thermo,USA)检测质量。
1.6 反转录及qRT-PCR分析
使用PrimeScriptTM Reagent Kit试剂盒(TaKaRa,RR037A)对样品RNA进行反转录;使用NCBI公共数据库网站在线引物设计(https://www.ncbi.nlm. nih.gov/tools/primer-blast/)qRT-PCR引物(表2),由上海生工生物工程有限公司合成;使用UltraSYBR Mixture(Low ROX)试剂盒(北京康为试剂生物科技有限公司,CW2601M)进行qRT-PCR检测。以作为内参基因,所有测试样品设置3次生物学重复,并通过2-△△Ct方法计算基因相对表达量,使用TBtools子程序HeatMap Illustrator绘制表达量热图。
2 结果
2.1 苦荞全基因组WOX基因成员序列特征
全基因组扫描分析,共鉴定出30个WOX基因,依据每个基因在苦荞染色体上的位置,依次暂命名为(图1)。30个WOX基因编码氨基酸数目范围为106—850 aa,预测相对分子量范围为12 596.49—93 358.70 Da,理论等电点范围为4.59—9.99。预测所有WOX基因的蛋白不稳定系数均大于40.0,推测为不稳定蛋白。19个WOX基因的蛋白二级结构主要以无规则卷曲为主,占比范围为42.28%—68.15%,其次为α螺旋、β转角和延伸链。11个WOX基因以α螺旋为主,占比范围为40.18%—53.00%,其次为无规则卷曲、β转角和延伸链。30个WOX基因均定位于细胞核内,无明显的信号肽特征信息出现。
聚类及结构域分析表明,30个WOX基因可聚为3类(Ⅰ、Ⅱ和Ⅲ),每一类具有独特的结构域、基序和结构特征。第Ⅰ类群有10个成员包含典型的HD及HALZ结构域,含1—2基序(motif),2—3个外显子。第Ⅱ类群有6个基因,包含HD、bZIP、START和MEKHLA结构域,均含有10个基序,9—18个外显子;但仅包含START和COG5576 superfamily结构域。剩余14个基因属于第Ⅲ类群仅含HB或HD结构域,含1—2基序,1—4个外显子(图2-A、图2-B和图2-C)。
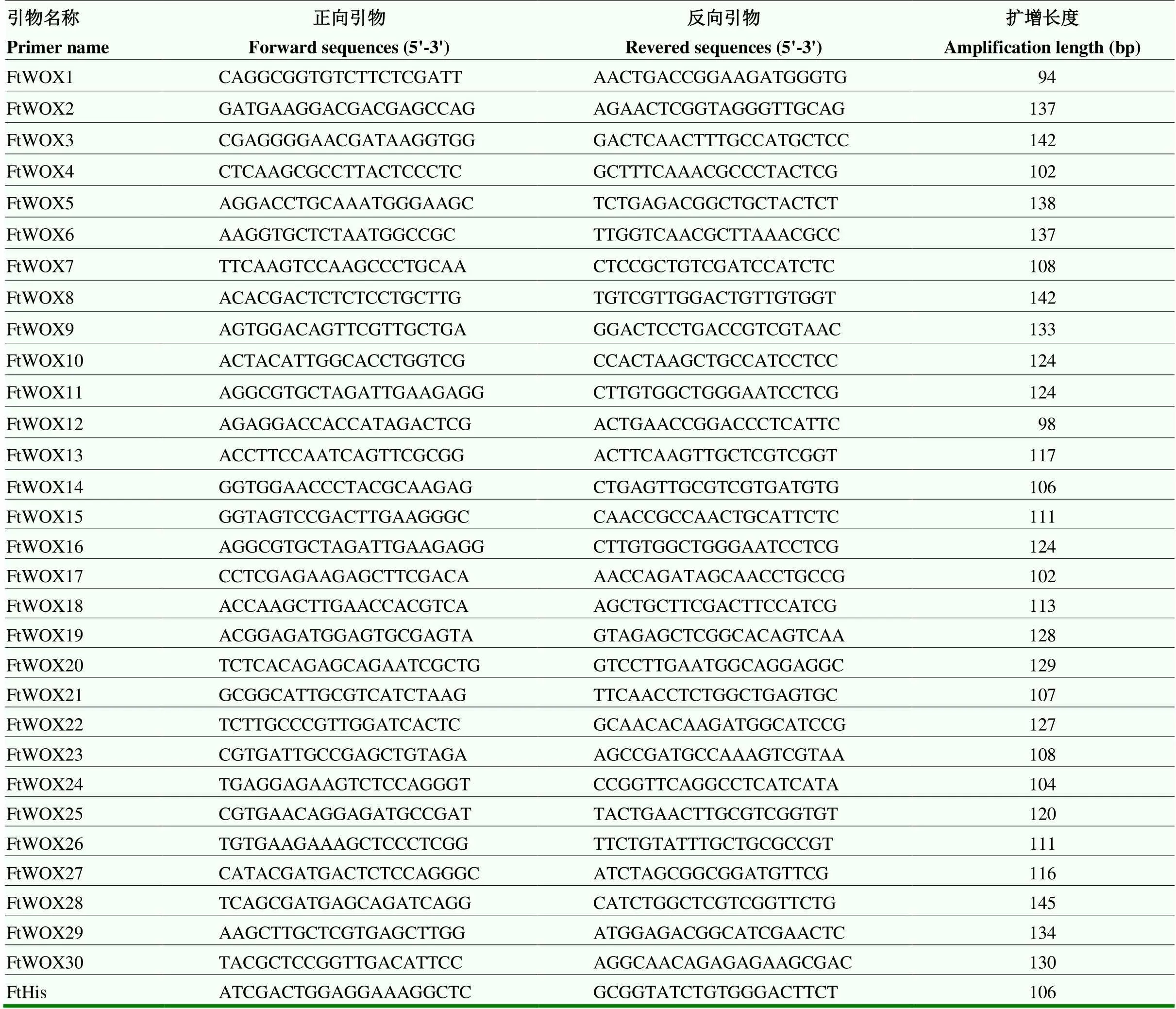
表2 实时荧光定量PCR引物序列
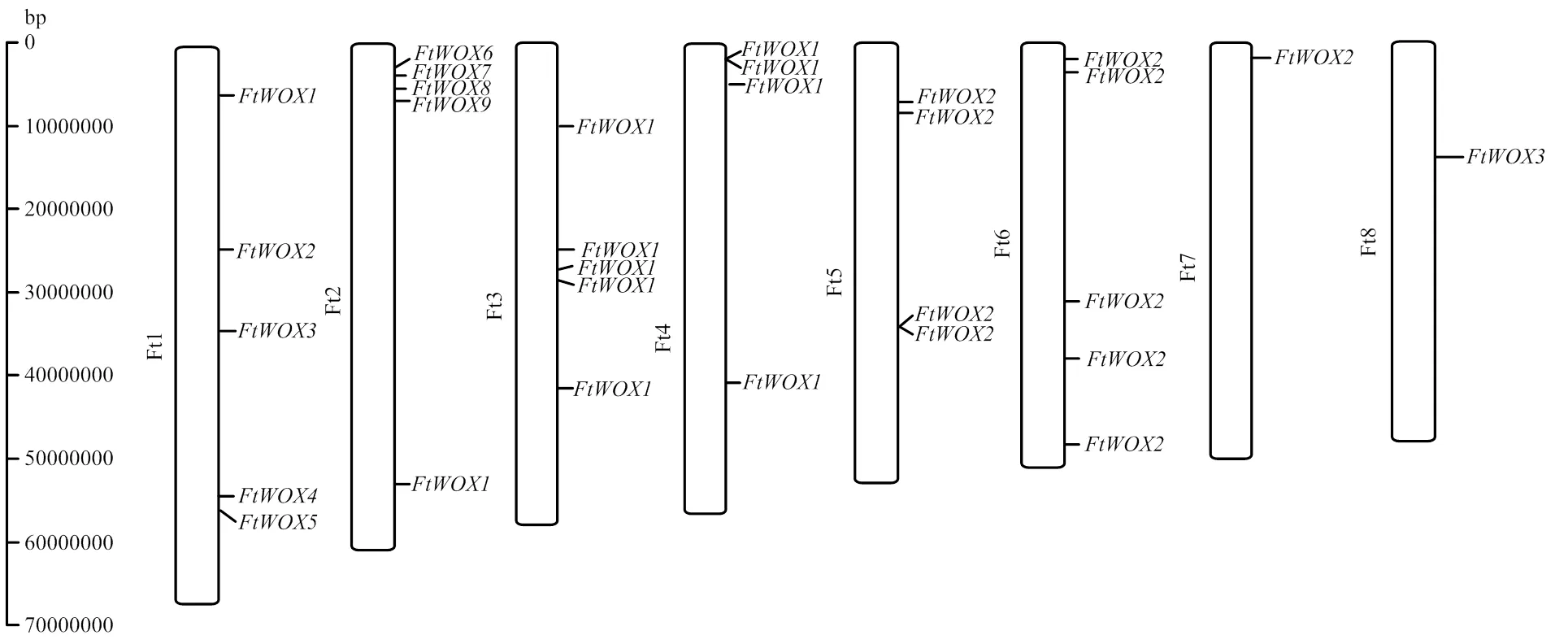
图1 苦荞WOX基因染色体定位图
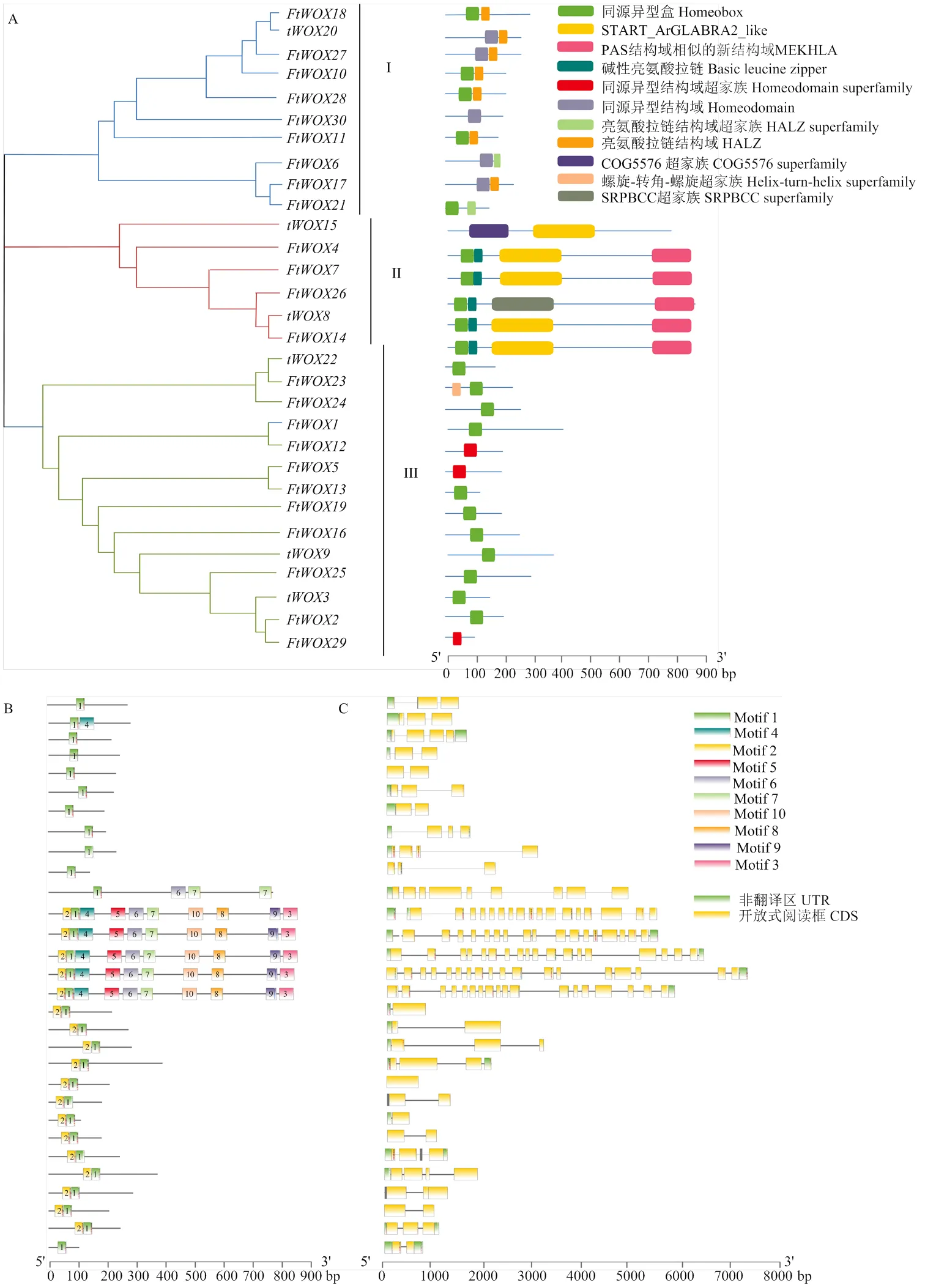
图2 苦荞FtWOXs基因家族系统进化及保守结构域(A)、保守基序(B)及基因结构(C)分析
系统进化树表明,来源于3个物种的57个WOX基因可以聚为3类。第1类共有15个基因,包含4个苦荞基因;第2类共26个基因,包括8个苦荞基因。值得注意的是,第3类16个基因全部来源于苦荞,表明这类苦荞WOX基因可能具有与拟南芥和水稻其他两类蛋白不同的功能特征(图3)。
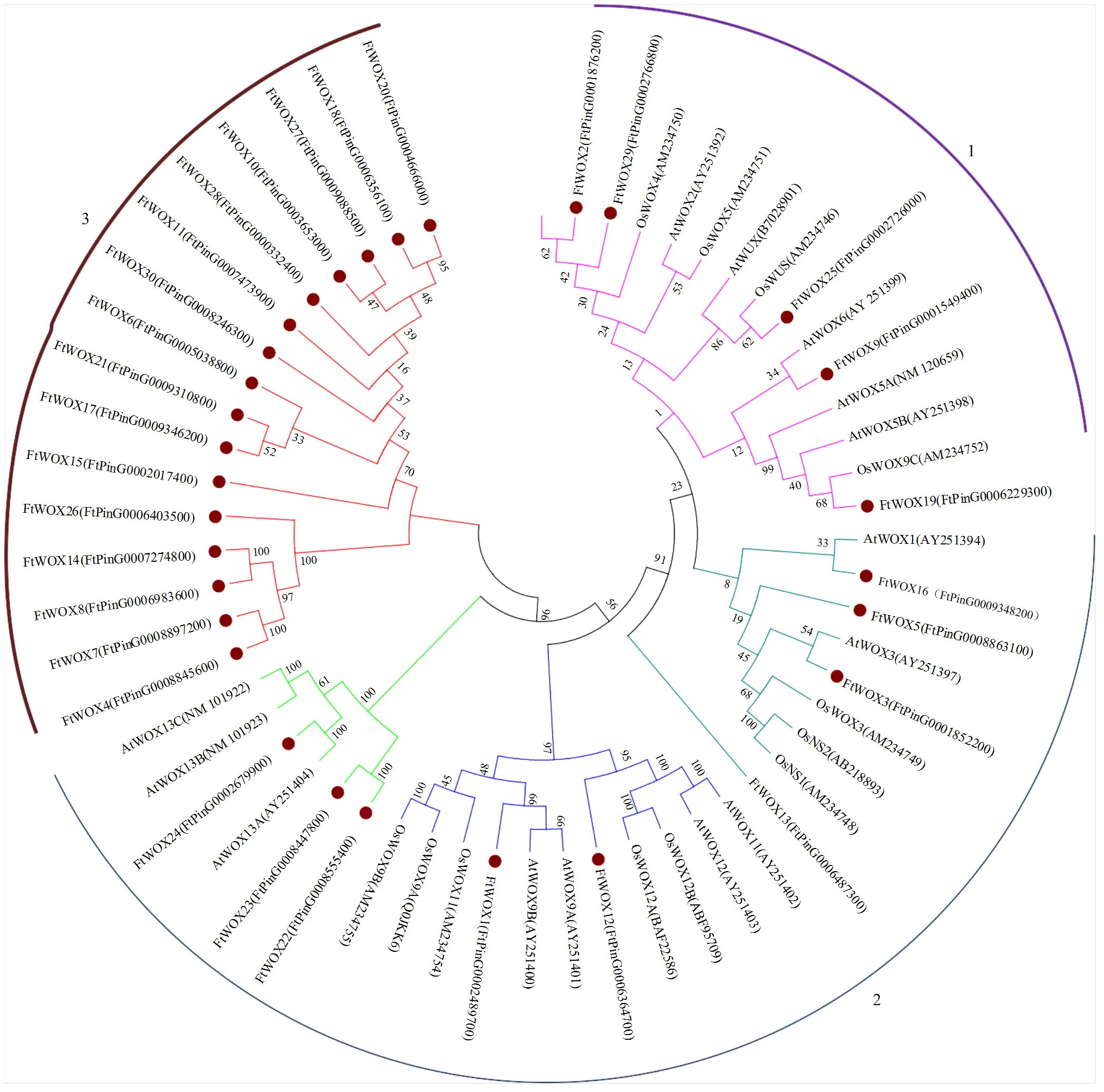
AtWOX1-13C和AtWUX:拟南芥WOX蛋白;OsWOX3-12B、OsWUX和OsNS1-2:水稻WOX蛋白;FtWOX1-30:苦荞WOX蛋白,括号内编号均为对应蛋白编号
30个WOX基因成员启动子序列,共预测到26个顺式作用元件,包含“分生组织形成相关”“光响应”“胚乳及种子发育”“赤霉素响应”“茉莉酸甲酯”及“MYB调控相关”等。其中4个WOX基因特有“分生组织形成相关”顺式作用元件。特有“胚乳发育”顺式作用元件。特有“种子发育”顺式作用元件(图4)。结果表明,一些WOX基因可能与胚乳或种子发育、分生组织形成密切相关。
与拟南芥共线性分析,发现6个FtWOX基因家族成员与拟南芥存在共线性关系,而其他24个基因均无共线性。2个物种共线性基因选择压力分析,发现这些基因Ka/Ks值<1,说明这6个WOX基因在苦荞和拟南芥之间是相对保守的,受到了纯化选择作用(图5)。

图4 苦荞FtWOXs基因启动子元件分析
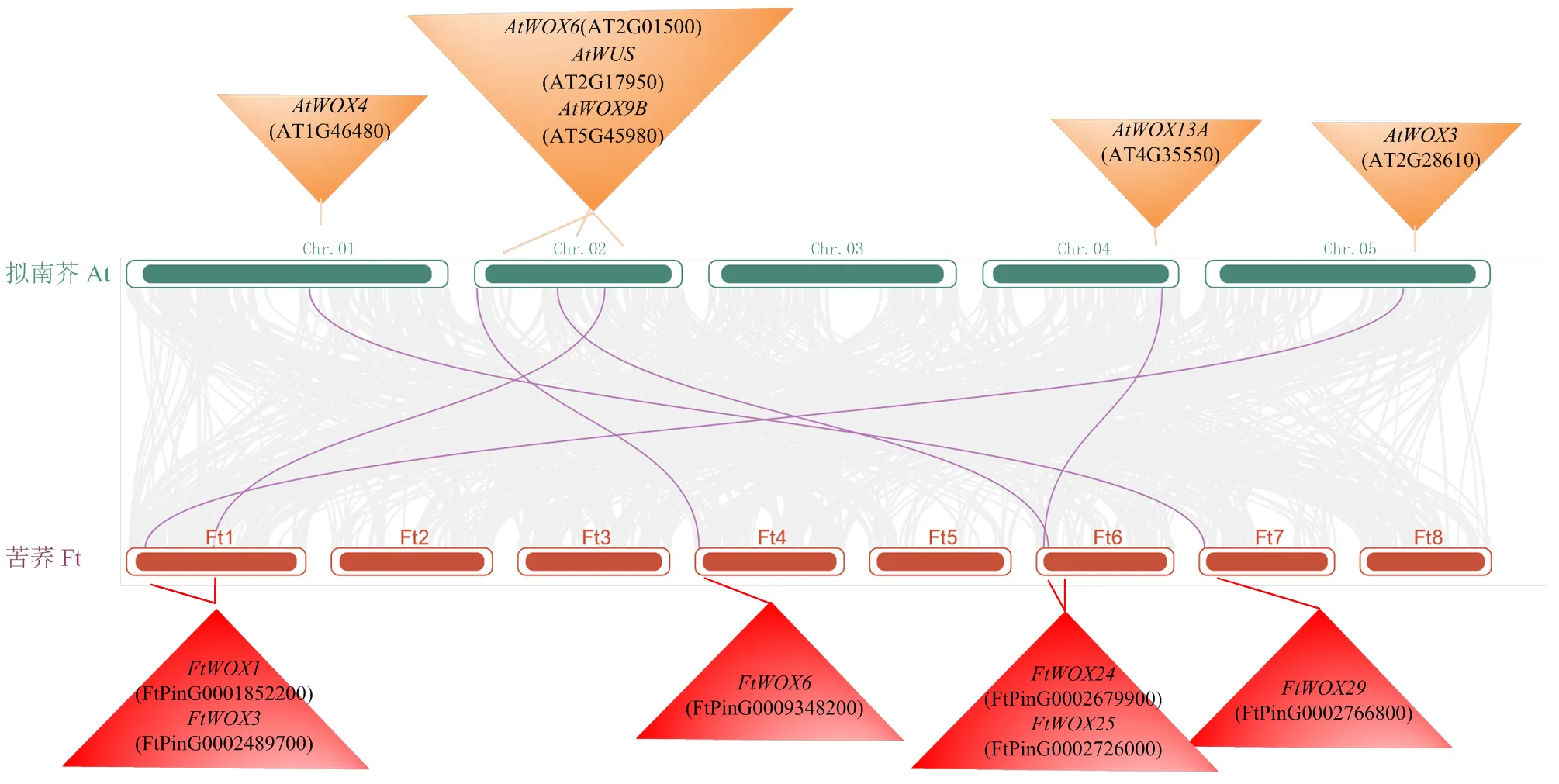
图5 苦荞(Ft)WOX基因家族与拟南芥(At)共线性分析
2.2 不同苦荞基因型出愈率评价
不同苦荞品种的愈伤组织状态及出愈率具有明显差异,如:PI658438品种出愈率达到了100%,且生长速度较快,愈伤组织颜色较绿,结构松散,可见绿色球状突起;而PI673865出愈率仅为20%,且愈伤组织生长速度慢,愈伤组织褐化严重(图6-A)。聚类分析结果显示,70份苦荞品种分为4类,第一类包含13个品种,出愈率高,介于92%—100%;第二类包含27个品种,出愈率介于66%—78%;第三类,有21个苦荞品种,出愈率较低,介于30%—64%;第四类的9个苦荞品种,出愈率均低于30%(图6-B)。
2.3 FtWOXs基因表达模式及与出愈率相关性分析
通过对30个FtWOXs基因在高、低出愈率品种愈伤组织中的表达模式进行分析,根据基因相对表达量可将FtWOXs基因分为2组,其中、和等8个基因为第I组,、和等22个基因为第II组。第I组的8个基因在低出愈率品种中高表达,而在高出愈率品种中低表达,如在ZNQ180(出愈率24%)中,相对表达量为16.71,而在PI673852(出愈率100%),该基因表达量为6.72。第II组的第3亚组包含了15个基因,均为在低出愈率品种中表达量较低,而在高出愈率品种中表达量较高(图7),如在5个低出愈率品种中表达值为2.03—5.35,而在5个高出愈率品种中表达值可达17.92—23.21。虽然上述基因相对表达量有一定的规律可循,但每个品种不同基因成员表达量差异较大。/与高、低出愈率之间关系最为明显,相关性分析结果显示,上述5个基因与苦荞出愈率呈极显著相关(<0.01),相关系数分别为0.908、0.897、0.894、0.882和0.876。结果表明,这5个WOX基因可作为高诱导愈伤组织基因型鉴定的标签基因,可进一步选择作为靶基因,通过基因工程的方法提高苦荞遗传转化效率(电子附表1)。
3 讨论
随着苦荞基因组的公布,对其功能基因及分子育种领域的研究需求越来越迫切[23]。苦荞具有严格的自花授粉特性,在基因功能及遗传机制研究方面具有优势。长期以来,对苦荞遗传转化体系的研究进程较为缓慢,主要研究集中在发根农杆菌转化体系的利用方面[24]。Hou等[25]利用苦荞和甜荞子叶再生体系研究了不同理化因子对芦丁含量影响,是较好的利用组织培养体系进行研究的例子。WOX基因被认为是保持茎尖分生组织分生能力的必需基因[26],因此,从WOX基因功能入手,打破苦荞再生困难瓶颈,使加快苦荞基因功能研究成为可能。

Ⅰ:出愈率为92%—100%的品种;Ⅱ:出愈率为66%—78%的品种;Ⅲ、Ⅳ:出愈率在30%—64%以及低于30%的品种
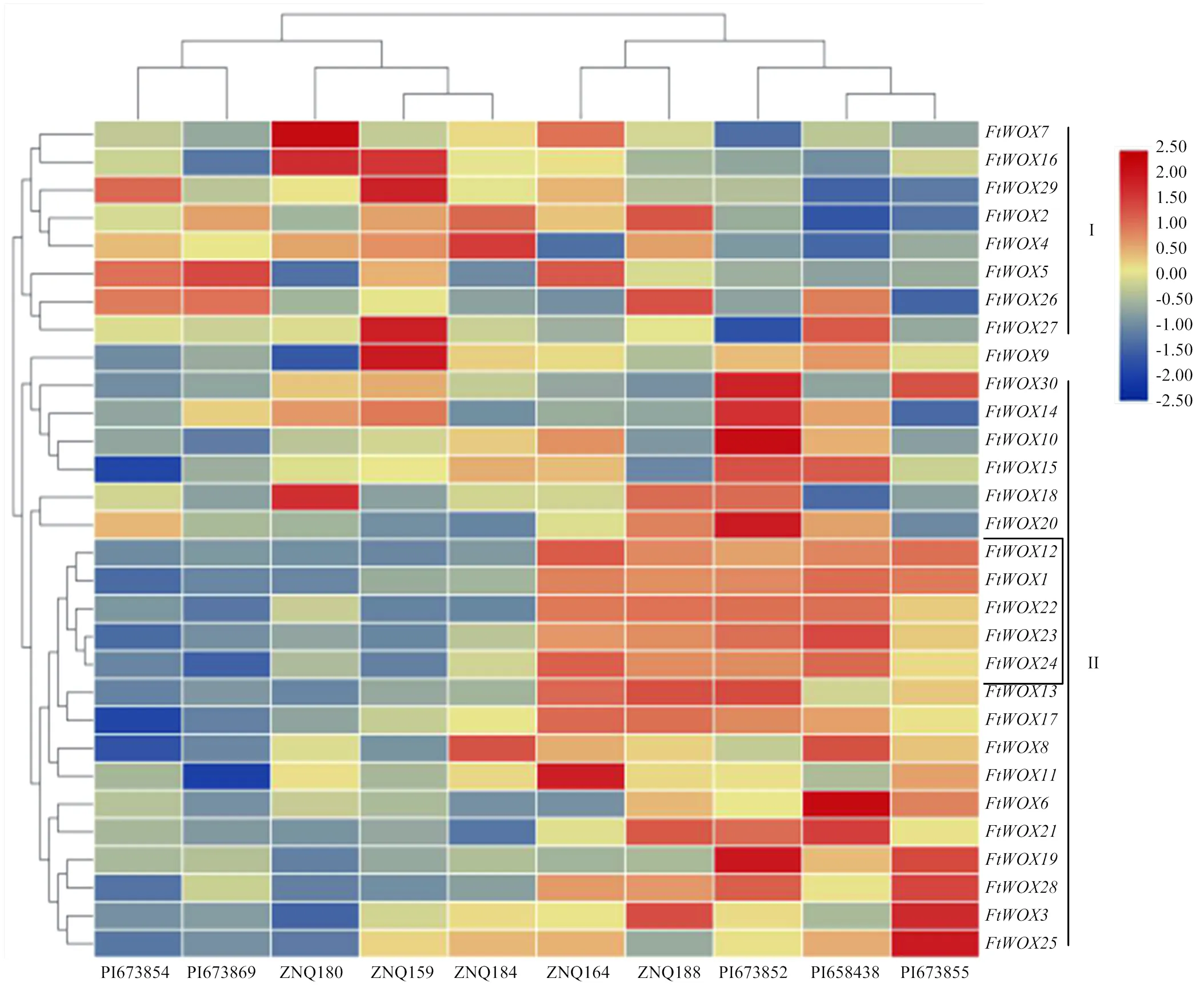
黑色框中5个基因表达为与出愈率高低具有一致性
本研究首次从全基因组水平扫描共获得30个苦荞WOX基因,这个数目约是拟南芥WOX基因数目个)和水稻(12个)的2倍[27],与大豆(33个)数目相当[28]。通过对其氨基酸序列保守基序及保守结构域分析表明,苦荞WOX基因成员均含有保守的motif1基序和HD结构域,而各成员间基因保守基序和结构域数目差异较大。值得注意的是,///这5个基因拥有最多的保守基序(motif1—motif10),也拥有最多的结构域,其中START和MEKHLA结构域,为HD-ZIP蛋白结构域,已有试验证据表明此类结构域与植物生长发育密切相关[29-30]。同时,研究发现WOX基因特有的HD结构域具有螺旋-转角-螺旋空间结构特征,属于DNA结合结构域,空间上与DNA的大沟和小沟结合从而调控基因的转录。同时也发现DNA结合结构域存在保守基序数目上的差别,这也进一步暗示了苦荞WOX基因家族成员功能的多样化[8]。
在拟南芥中,不同的WOX基因成员执行不同的功能,系统进化树研究为预测基因功能提供了参考。基因结构是基因组进化和基因功能变异的重要证据,在本研究中,30个WOX基因中有5个与//等基因聚为一类,具有典型的HD结构域,被认为属于‘modern clade’[27,31];而单独聚为一类的16个苦荞WOX基因发现与拟南芥保守基序上有较大差异,表明其功能上的分化。
对30个WOX基因启动子序列分析共预测到26个顺式作用元件,其中发现大量与激素调控相关的顺式作用元件,此结果证实了生长素、细胞分裂素、赤霉素、脱落酸等激素的顺式作用元件广泛存在于WOX基因的启动子区域,说明WOX家族基因参与激素调控从而影响植株分生能力。此外,预测到了可能与胚乳或种子发育、分生组织形成密切相关。
不同基因型苦荞的出愈率有明显的差异性,这一结果在水稻、小麦和苦荞愈伤组织的诱导中均有体现[32-34],而WOX基因的表达水平影响着植物组织培养中外植体愈伤组织再生的能力[35]。本研究发现5个FtWOX基因在10个出愈率显著差异的苦荞愈伤组织中表达模式与苦荞的出愈率呈显著正相关关系,进一步表明WOX基因对苦荞出愈率的影响,推测在苦荞中过表达WOX基因可能会影响苦荞外植体产生愈伤组织的能力。拟南芥中愈伤组织或根源基的形成需要激活生长素响应因子ARF7/ARF19,从而启动下游的WOX11-LBD16/17/18/29-WOX5信号通路。另外,拟南芥中是不定根生长发育的关键调节基因[36],该基因编码的蛋白序列与和的蛋白同源性较高,而这两个基因表达均与苦荞出愈率密切相关,这也进一步暗示了苦荞中部分WOX基因在愈伤组织诱导中起到重要作用[37]。此外,//聚类分析显示其与和处在同一分支,作为PXY途径的下游元件调节形成层细胞的分化[38-39]。这些结果同样暗示苦荞//与出愈率相关性,可能与其启动子中含有生长素相关的顺式作用元件或对细胞分化的调节有关。因此,推测和//等基因可能是提高苦荞愈伤组织出愈率的关键靶点,可通过对基因表达情况的检测来确定不同品种出愈率的情况,从而为筛选高出愈率品种提供新的快速检测方法。
4 结论
全基因组范围内共鉴定出30个苦荞WOX基因,不均匀分布于8条染色体上,亚细胞定位于细胞核,具有典型的HD结构域特征,与拟南芥6个WOX基因具有较高同源性及基因组共线性,其中16个WOX基因为苦荞特有成员。70个苦荞品种表现出不同的愈伤组织诱导率。WOX基因表达水平与不同品种出愈率之间存在正相关关系。
[1] JOSHI D C, CHAUDHARI G V, SOOD S, KANT L, PATTANAYAK A, ZHANG K X, FAN Y, JANOVSKA D, MEGLIC V, ZHOU M L. Revisiting the versatile buckwheat: reinvigorating genetic gains through integrated breeding and genomics approach. Planta, 2019, 250: 783-801.
[2] Giménez-Bastida J A, Zieliński H. Buckwheat as a functional food and its effects on health.Journal of Agricultural and FoodChemistry, 2015, 63(36): 7896-7913.
[3] CAMPBELL C. Buckwheat crop improvement. Fagopyrum, 2003, 20: 1-6.
[4] 王兴春, 李宏, 王敏, 杨致荣. 植物体细胞胚胎发生的调控网络. 生物工程学报, 2010, 26(2): 141-146.
WANG X C, LI H, WANG M, YANG Z R. Regulatory networks of somatic embryogenesis in plant. Chinese Journal of Biotechnology, 2010, 26(2): 141-146. (in Chinese)
[5] GENRING W J, AFFOLTER M, BURGLIN T. Homeodomain proteins. Annual Review of Biochemistry, 1994, 63: 487-526.
[6] DERELLE R, LOPEZ P, GUYADER H L, MANUEL M. Homeodomain proteins belong to the ancestral molecular toolkit of eukaryotes. Evolution & Development, 2007, 9(3): 212-219.
[7] LAUX T, MAYER K F, BERGER J, JURGENS G. Thegene is required for shoot and floral meristem integrity in. Development, 1996, 122(1): 87-96.
[8] GRAAFF E, LAUX T, RENSING S A. The WUS homeobox- containing (WOX) protein family. Genome Biology, 2009, 10: 248.
[9] ISABEL B, THOMAS L. Regulation of WUSCHEL transcription in the stem cell niche of theshoot meristem. The Plant Cell, 2005, 17(8): 2271-2280.
[10] MENG W J, CHENG Z J, SANG Y L, ZHANG M M, RONG X F, WANG Z W, TANG Y Y, ZHANG X S. Type-Bresponse regulators specify the shoot stem cell niche by dual regulation of WUSCHEL. The Plant Cell, 2017, 29(6): 1357-1372.
[11] ZUO J R, NIU Q W, GIOVANNA F, CHUA N H. Thegene promotes vegetative to embryonic transition in. The Plant Journal, 2002, 30(3): 349-359.
[12] Gambino G, Minuto M, Boccacci P, Perrone I, Vallania R, Gribaudo I. Characterization of expression dynamics of WOX homeodomain transcription factors during somatic embryogenesis in. Journal of Experimental Botany, 2011, 62(3): 1089-1101.
[13] NARDMANN J, WERR W. The invention of WUS-like stem cell-promoting functions in plants predates leptosporangiate ferns. Plant molecular biology, 2012, 78: 123-134.
[14] BREUNINGER H, RIKIRSCH E, HERMANN M, UEDA M, LAUX T. Differential expression of WOX genes mediates apical- basal axis formation in theembryo. Cell, 2008, 14(6): 867-876.
[15] ZHU J, SHI H, LEE B H, DAMSZ B, CHENG S, STIRM V, ZHU J K, HASEGAWA P M, BRESSAN R A. Anhomeodomain transcription factor gene, HOS9, mediates cold tolerance through a CBF-independent pathway. Proceedings of the National Academy of Sciences of the United States of America, 2004, 101(26): 9873-9878.
[16] CHENG S, HUANG Y, ZHU N, ZHAO Y. The rice WUSCHEL- related homeobox genes are involved in reproductive organ development, hormone signaling and abiotic stress response. Gene, 2014, 549(2): 266-274.
[17] MARCHLER-BAUER A, BO Y, HAN L Y, HE J, LANCZYCKI C J, LU S, CHITSAZ F, DERBYSHIRE M K, GEER R C, GONZALES N R, GWADZ M, HURWITZ D I, LU F, MARCHLER G H, SONG J S, THANKI N, WANG Z X, YAMASHITA R A, ZHANG D C, ZHENG C J, GEER L Y, BRYANT S H. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic acids research, 2017, 45(D1): 200-203.
[18] CHEN C J, Chen H, ZHANG Y, THOMAS H R, FRANK M H, HE Y H, XIA R. TBtools:An integrative toolkit developed for interactive analyses of big biological data. Molecular Plant, 2020, 13(8): 1194-1202.
[19] CHENNE R, SUGAWARA H, KOIKE T, LOPEZ R, GIBSON T J, HIGGINS D G, THOMPSON J D. Multiple sequence alignment with the clustal series of programs. Nucleic acids research, 2003, 31(13): 3497-3500.
[20] KUMAR S, STECHER G, LI M, KNYAZ C, TAMURA K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Molecular biology and evolution, 2018, 35(6): 1547-1549.
[21] BAILEY T L, BODEN M, BUSKE F A, FRITH M, GRANT C E, CLEMENTI L, REN J Y, LI W, NOBLE W S. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Research, 2009, 37(suppl 2): 202-208.
[22] LESCOT M, DEHAIS P, THIJS G, MARCHAL K, MOREAU Y, VAN D P Y, ROUZE P, ROMBAUTS S. PlantCARE, a database of plant-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Research, 2002, 30(1): 325-327.
[23] ZHANG L J, LI X X, MA B, GAO Q, DU H L, HAN Y H, LI Y, GAO Y H, QI M, ZHU Y X, LU H W, MA M C, LIU L L, ZHOU J P, NAN C H, QIN Y J, WANG J, GUI L, LIU H M, LIANG C Z, QIAO Z J. The tartary buckwheat genome provides insights into rutin biosynthesis and abiotic stress tolerance. Molecular plant, 2017, 10(9): 1224-1237.
[24] MI Y L, ZHU Z H, QIAN G T, LI Y, MENG X X, XUE J P, CHEN Q F, SUN W, SHI Y H. Inducing hairy roots by Agrobacterium rhizogenes-mediated transformation in tartary buckwheat (). Journal of visualized experiments, 2020(157): e60828.
[25] HOU S Y, SUN Z X, LINGHU N, WANG Y G, HUANG K S, XU D M, HAN Y H. Regeneration of buckwheat plantlets from hypocotyl and the influence of exogenous hormones on rutin content and rutin biosynthetic gene expression. Plant Cell, Tissue and Organ Culture, 2015, 120(3): 1159-1167.
[26] MAYER K F, SCHOOF H, HAECKER A, LENHARD M, JURGENS G, LAUX T. Role of WUSCHEL in regulating stem cell fate in theshoot meristem. Cell, 1998, 95(6): 805-815.
[27] WU C C, LI F W, KRAMER E M. Large-scale phylogenomic analysis suggests three ancient superclades of the WUSCHEL-related homeobox transcription factor family in plants. PLoS ONE, 2019, 14(10): e0223521.
[28] HAO Q, ZHANG L, YANG Y, SHAN Z, ZHOU X A. Genome wide analysis of the WOX gene family and function exploration of
[29] VENKATA B, SCHRICK K. START Domains in Lipid/Sterol Transfer and Signaling in Plants. Michigan State University: Michigan State University Press, 2006.
[30] MUKHERJEE K, BURGLIN T R. MEKHLA, a novel domain with similarity to PAS domains, is fused to plant homeodomain-leucine zipper III proteins. Plant Physiology, 2006, 140(4): 1142-1150.
[31] ZHANG X, ZONG J, LIU J H, YIN J Y, ZHANG D B. Genome-wide analysis of WOX gene family in rice, sorghum, maize,and poplar.Journal of Integrative Plant Biology,2010, 52(11): 1016-1026.
[32] HOQUE M E, MANSFIELD J W. Effect of genotype and explant age on callus induction and subsequent plant regeneration from root derived callus of indica rice genotypes. Plant Cell Tissue and Organ Culture, 2004, 78(3): 217-223.
[33] 张小红, 闵东红, 邵景侠. 小麦愈伤组织诱导及原生质体的分离与纯化. 中国农学通报, 2010, 26(21): 49-53.
ZHANG X H, MIN D H, SHAO J X. Wheat callus induction and protoplasts of separation and purification. China Agricultural Journal, 2010, 26(21): 49-53. (in Chinese)
[34] 王鹏姬. 荞麦愈伤组织培养及其黄酮合成研究[D]. 杨凌: 西北农林科技大学, 2013.
WANG P J. Research on callus culture and flavonoids biosynthesis of buckwheat[D]. Yangling: Northwest A&F University, 2013. (in Chinese)
[35] LIU B L, WANG L, ZHANG J, LI J B, ZHENG H Q, CHEN J, LU M Z. WUSCHEL-related homeobox genes in: diversified expression patterns and a functional similarity in adventitious root formation. BMC Genomics, 2014, 15: 296.
[36] GUO F, ZHANG H, LIU W, HU X, HAN N, QIAN Q, XU L, BIAN H. Callus initiation from root explants employs different strategies in rice and. Plant Cell Physiology, 2018, 59(9): 1782-1789.
[37] Deveaux Y, Toffano-Nioche C, Claisse G, Thareau V, Morin H, Laufs P, Moreau H, Kreis M, Lecharny A. Genes of the most conserved WOX clade in plants affect root and flower development in. BMC Evolutionary Biology, 2008, 8: 291.
[38] Hirakawa Y, Kondo Y, Fukuda H. TDIF peptide signaling regulates vascular stem cell proliferation via the WOX4 homeobox gene in. The Plant Cell, 2010, 22(8): 2618-2629.
[39] ETCHELLS J P, PROVOST C M, MISHRA L, TURNER S. WOX4 and WOX14 act downstream of the PXY receptor kinase to regulate plant vascular proliferation independently of any role in vascular organization. Development, 2013, 140: 2224-2234.
Genome-Wide identification of WOX family and expression analysis of callus induction rate in Tartary buckwheat
HOU SiYu1, WANG XinFang1, DU Wei1, FENG JinHua1, HAN Yuanhuai1, LI HongYing1, LIU LongLong2, SUN ZhaoXia1
1College of Agronomy, Shanxi Agricultural University, Taigu 030801, Shanxi;2Center for Agricultural Genetic Resources Research, Shanxi Agricultural University, Taiyuan 030031
【】This study aimed to identify the whole genome WOX (WUSCHEL-related home obox) gene family in Tartary buckwheat and reveal the correlation with sequence characteristics of its gene family members, gene expression pattern and the rate of callus induction. It provides a theoretical basis for breaking through the regeneration and genetic transformation problem of Tartary buckwheat.【】The protein and nucleic acid sequence of the WOX gene family members in Tartary buckwheat were obtained by homology blast and the sequence ofWOX genes were served as reference. Based on protein homology and conserved domain analysis, all members of Tartary buckwheat WOX gene family were identified. The TBtools software was used to further demonstrate the characteristics of the WOX genes in Tartary buckwheat, including gene structure, conserved domain and-acting element. Genomic collinearity of WOX gene family members between Tartary buckwheat andwas analysed. Based on proximity method, the MEGA X softwarewas used to perform phylogenetic tree of these WOX genes in Tartary buckwheat,and rice. The hypocotyl explants of 70 Tartary buckwheat varieties were cultured with MS+2,4-D 3.0 mg·L-1+6-BA 1.0 mg·L-1for callus induction and the callus emergence rate of different genotypes was evaluated. The FtWOX gene expression level was performed by qPCR to compare the different Tartary buckwheat varieties with high and low callus yield. The correlation between callus rate and FTWOXS gene family members was analysed based on Pearson correlation coefficient. 【】 A total of 30 WOX genes were identified in Tartary buckwheat and they were unevenly distributed on 8 chromosomes. The 30 Tartary buckwheat WOX genes could be divided into three groups by phylogenetic tree. The WOX genes contained different conserved domains in different groups, and the main conserved domains were HD(Homeodomain), START and Mekhla. The conserved motif analysis showed that the conserved motif number ofgenes may contain 2 to 10 motifs, and the gene structure analysis showed that the number of exons contained in the genes between 2 to 18. Promoter elements analysis showed 26 different kinds of-acting elements in the 30 WOX genes. The phylogenetic analysis showed that 30 Tartary buckwheat, 15and 12 rice WOX gene family members could be divided into three categories, of which the third group is unique to Tartary buckwheat. The collinearity analysis showed that six WOX genes were genomic collinearity between Tartary buckwheat and. Expression pattern and correlation analysis show that the expression level of////has positive correlation with the callus induction. 【】 Collectively, these data suggest that the Tartary buckwheat FtWOX members showed abundant sequence variation characteristics. The expression level and callus rate of WOX gene in different Tartary buckwheat genotypes were significantly different and correlated to some extent, suggesting that different Tartary buckwheat WOX genes had potential functional diversity.
Tartary buckwheat; WOX gene family; callus induction; gene expression

10.3864/j.issn.0578-1752.2021.17.002
2021-02-18;
2021-05-08
国家重点研发计划中欧政府间合作项目(2017YFE0117600)、国家自然科学基金(32070365)、山西省农业科学院应用基础研究计划(YGC2019FZ2)、财政部和农业农村部:国家现代农业产业技术体系(CARS-07-A-2)
侯思宇,Tel:18635068055;E-mail:bragren123@163.com。通信作者孙朝霞,Tel:18636071356;E-mail:18636071356@163.com
(责任编辑 李莉)
猜你喜欢
福建农林大学学报(自然科学版)(2022年4期)2022-11-01
南方医科大学学报(2022年3期)2022-04-13
农业科技与信息(2021年8期)2021-12-06
诗潮(2021年11期)2021-11-24
三农资讯半月报(2020年15期)2020-08-25
飞碟探索(2015年9期)2015-11-05
红领巾·探索(2015年9期)2015-09-10
农村农业农民·B版(2014年12期)2015-01-04
恋爱婚姻家庭·养生版(2011年8期)2011-05-14
