
酚类菊糖衍生物的制备及抗氧化活性
2021-10-31 11:19谭文强郭占勇
食品科学 2021年19期
李 青,谭文强,苗 芹,郭占勇
(中国科学院烟台海岸带研究所,海岸带生物学与生物资源利用重点实验室,山东 烟台 264003)
菊糖,又名菊粉,具有调节肠胃功能、提高免疫力及促进矿物质吸收等多种保健作用,可以用作食品添加剂和膳食补充剂,已有40多个国家将其批准为功能食品,并广泛应用到医药、保健品、食品工业等领域[1-3]。在我国菊糖也正在被大众所认知,菊糖生产及其相关行业迅速增长。菊糖具有一定的生物活性,作为一类可再生、无毒、生物相容性好的天然多糖,来源丰富,结构相对简单,且有可修饰的羟基,具有较大的结构修饰空间。因此,通过化学修饰对菊糖进行结构改性提高其生物性能的相关研究备受关注。目前在天然多糖化学修饰的相关研究中,壳聚糖和甲壳素等报道较多[4-8],而针对菊糖的结构改性及相应的产品开发相对较少。有文献报道在菊糖上连接咪唑环并季铵化后,菊糖衍生物质量浓度为1.6 mg/mL时对超氧阴离子自由基(O-2·)和羟自由基(·OH)的清除率分别为67.8%和86.7%[9],与菊糖相比有大幅度提高。本课题组前期研究[10-11]发现,在多糖上连接活性基团可以提高其抑菌性和抗氧化活性。连接三氮唑鎓盐的壳聚糖衍生物和菊糖衍生物都表现出很高的清除能力(半抑制质量浓度(half maximal inhibitory concentration,IC50)<0.01 mg/mL),与抗坏血酸相近。由此可见,通过化学修饰提高菊糖的抗氧化活性有非常大的潜力。
近些年,氧化应激引起的健康问题受到很大的重视,其诱导机体衰老并加剧阿尔茨海默病、心血管疾病、恶性肿瘤等疾病[12]。抗氧化剂可以清除过量的自由基减轻机体的氧化胁迫损伤,从而有助于延缓衰老,保护人体免受多种慢性疾病的侵袭[13];另一方面能够有效保证药品品质并延长有效期,因此受到广泛的关注,被食品、保健品、化妆品企业列为重要研发内容[14-15]。自由基清除能力是抗氧化能力的重要指标,菊糖本身就有一定的自由基清除能力,而且具有无毒、无刺激、生物相容性好、来源丰富、价格低廉和易修饰等优点,是开发天然抗氧化剂的优良选择[11,16]。
为开发菊糖衍生物的实际应用性,本实验选择兼具高生物活性和安全性的多酚类化合物,对菊糖进行化学修饰。酚类化合物具有抗氧化、抗癌、抗炎和抗过敏等活性[17]。有些酚类化合物在抗氧化、降血脂等方面有很强的药理作用,被广泛用于抗动脉粥样硬化、抗肿瘤、预防阿尔茨海默症、抑制血小板聚集等方面[18-21]。基于此,在前期研究基础上制备一系列共6 种酚基菊糖衍生物,并测定其抗氧化活性,以期为开发菊糖衍生物在抗氧化剂或自由基清除剂等方面的应用提供依据。
1 材料与方法
1.1 材料与试剂
菊糖(分子质量8 000~12 000 Da) 西安百川生物科技有限公司;芳香醛(4-羟基苯甲醛、3-羟基苯甲醛、3,4-二羟基苯甲醛、2,3-二羟基苯甲醛、3-甲氧基-4-羟基苯甲醛和3,5-二甲氧基-4-羟基苯甲醛,纯度≤98%)默克生命科学(上海)有限公司;碘甲烷、碘化钠、氢氧化钠、N-溴丁亚胺、三苯基膦、溴化钾、乙二胺均为分析级 国药集团化学试剂(上海)有限公司。
1.2 仪器与设备
IS50傅里叶变换红外光谱(Fourier transform infrared spectroscopy,FTIR)仪 上海赛默飞科技有限公司;AVIII-500核磁共振(nuclear magnetic resonance,NMR)光谱仪 瑞士Bruker公司;DNM-9602G酶标仪 北京普朗新技术有限公司。
1.3 方法
1.3.1 菊糖衍生物的合成
菊糖衍生物的合成路线如图1所示。
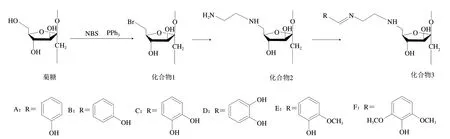
图1 菊糖衍生物制备路线Fig. 1 Synthetic routes for the preparation of inulin derivatives
溴代菊糖(化合物1)的合成:按照文献[22]中所述方法进行。以N-溴代丁二酰亚胺(N-bromosuccinimide,NBS)为溴代试剂与菊糖C6位伯羟基反应合成溴代菊糖(化合物1)。
化合物2的合成:在室温下取2 mmol化合物1加入5 mL乙二胺中,在60 ℃搅拌反应16 h后将混合物倒入过量无水乙醇中,过滤沉淀,用无水乙醇反复洗涤,收集沉淀,冷冻干燥得6-N-(氨基乙基)-6-脱氧菊糖(化合物2)。
化合物3的合成:将1.0 mmol化合物2与1.5 mmol羟基苯甲醛(4-羟基苯甲醛、3-羟基苯甲醛、3,4-二羟基苯甲醛、2,3-二羟基苯甲醛、2-甲氧基-3-羟基苯甲醛和2,4-二甲氧基-3-羟基苯甲醛)加入10 mL二甲基亚砜中,75 ℃氩气保护下搅拌20 h,然后将混合物倒入过量无水乙醇中,过滤沉淀,用乙醇反复洗涤。产物在索氏提取器中用无水乙醇提取48 h,产物在60 ℃下干燥24 h。
1.3.2 菊糖及其衍生物的FTIR和1H NMR分析
取一定量菊糖及1.3.1节制备的菊糖衍生物进行FTIR分析,KBr压片,扫描范围为4 000~500 cm-1。对1.3.1节制备的菊糖衍生物进行1H NMR分析,频率500 MHz,溶剂为氘代二甲基亚砜(dimethyl sulfoxide-d6,DMSO-d6)。
1.3.3 抗氧化活性测定1
按照文献[23]的方法进行,并稍加修改。用蒸馏水配制质量浓度分别为0.02、0.04、0.2、0.4、0.8 mg/mL的样品溶液。·在吩嗪硫酸甲酯(phenazine methosulfate,PMS)-烟酰胺腺嘌呤二核苷酸(nicotinamide ad enine dinucleotide,NADH)体系中产生。用Tris-HCl缓冲液(16 mmol/L、pH 8.0)分别配制NADH(365.7 μg/mL)、硝基蓝四氮(245.3 μg/mL)和PMS(18.38 μg/mL)溶液。分别取0.5 mL NADH、PMS、硝基蓝四氮溶液与不同质量浓度样品溶液混合,使混合溶液中样品终质量浓度分别为0.005、0.01、0.05、0.1、0.2 mg/mL。按以上方法配制混合溶液,以不加样品溶液为空白组,不加NADH的溶液为对照组。室温反应5 min,于560 nm波长处测定溶液吸光度(A560nm)。清除率按式(1)计算。
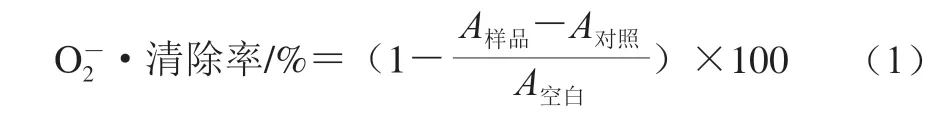
式中:A样品表示样品组溶液A560nm;A对照表示对照组溶液A560nm;A空白表示空白组溶液A560nm。
1.3.3.2 ·OH清除能力
·OH清除能力按照文献[24]的方法进行,并稍加修改。用蒸馏水配制质量浓度分别为0.4、0.8、1.2、1.6、2.4 mg/mL的样品溶液,用磷酸盐缓冲溶液(pH 7.4)配制体积分数3% H2O2溶液、360 μg/mL番红花T溶液和2 mmol/L乙二胺四乙酸-Fe2+溶液。分别取0.25 mL乙二胺四乙酸-Fe2+溶液、0.5 mL磷酸盐缓冲溶液、0.5 mL番红花T溶液、0.5 mL H2O2溶液与0.25 mL不同质量浓度样品溶液混合,使混合溶液中样品终质量浓度分别为0.05、0.1、0.15、0.2、0.3 mg/mL。按以上方法配制混合溶液,以不加样品溶液作为空白组,不加样品溶液和H2O2溶液作为对照组。37 ℃反应30 min,于520 nm波长处测定溶液吸光度(A560nm)。·OH清除率按式(2)计算。

式中:A样品表示样品组溶液A560nm;A对照表示对照组溶液A560nm;A空白代表空白组溶液A560nm。
1.3.3.3 DPPH自由基清除能力
DPPH自由基清除能力按照文献[25]的方法进行。用蒸馏水配制质量浓度分别为0.005、0.01、0.02、0.1、0.2 mg/mL的样品溶液,用无水乙醇配制180 μmol/L DPPH溶液。分别取1 mL DPPH溶液和1 mL不同质量浓度样品溶液混合,使混合溶液中样品终质量浓度分别为0.002 5、0.005、0.01、0.05、0.1 mg/mL。按以上方法配制混合溶液,以不加样品溶液作为空白组,用乙醇代替DPPH溶液作为对照组。室温反应30 min,测定517 nm波长处溶液吸光度(A517nm)。DPPH自由基清除率按式(3)计算。

式中:A样品表示样品组溶液A517nm;A对照表示对照组溶液A517nm;A空白代表空白组溶液A517nm。
1.3.3.4 还原能力
按照文献[26]的方法进行,并稍加修改[26]。用蒸馏水配制质量浓度分别为0.03、0.17、0.33、0.67、1 mg/mL的样品溶液。分别取0.3 mL不同质量浓度样品溶液与0.3 mL 1 g/100 mL铁氰化钾溶液混合,50 ℃反应20 min,然后加入0.3 mL体积分数10%三氯乙酸溶液终止反应,离心后取上清液加入0.1 mL 0.1 g/100 mL氯化铁溶液,室温反应10 min后,于700 nm波长处测定溶液的吸光度(A700nm)。用吸光度表示还原能力。
1.3.3.5 Fe2+螯合能力
Fe2+螯合能力按照文献[26]方法进行测定。分别取1.8 mL无水甲醇、0.05 mL 2 mmol/L氯化亚铁溶液、0.1 mL 5.0 mmol/L菲洛嗪溶液与0.05 mL不同质量浓度(0.04、0.4、2、4、6 mg/mL)的样品溶液混合,使混合溶液中样品终质量浓度分别为0.001、0.01、0.05、0.1、0.15 mg/mL。按以上方法配制混合溶液,以不加样品溶液作为空白组。室温反应10 min,于562 nm波长处测定溶液吸光度(A562nm)。Fe2+螯和率按式(4)计算。
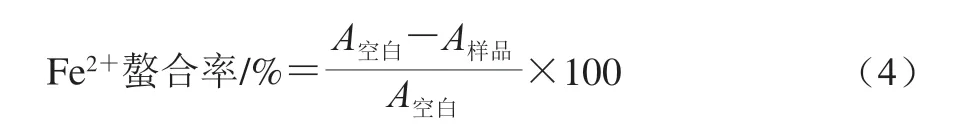
式中:A样品表示样品组溶液A562nm;A空白表示空白组溶液A562nm。
抗氧化活性指标(还原能力除外)均以IC50表示。
1.3.4 细胞毒性测定
以L929细胞为模型,对菊糖及其衍生物进行细胞毒性测定。采用CCK-8法进行体外细胞毒性测定[23]。L929细胞培养至指数生长期,配制为浓度1.0×105个/mL细胞悬液,接种于96 孔板,每孔100 μL。37 ℃、5% CO2、饱和湿度培养箱中培养24 h后,每孔加入不同质量浓度(31.25、62.5、125、250 μg/mL)的菊糖及其衍生物溶液100 μL,每个质量浓度3~5 个复孔,继续培养24 h后,离心,吸取上层清液,每孔加入100 μL CCK-8试剂,37 ℃下继续培养4 h,用酶标仪于450 nm波长处测定溶液吸光度(A450nm)。用等体积蒸馏水代替样品溶液作为对照组。细胞存活率按式(5)计算。

式中:A样品代表样品组A450nm;A对照代表对照组A450nm。
细胞培养方法同上。使用不同质量浓度的菊糖衍生物化合物3C处理L929细胞24 h后,用普通光学显微镜在放大20 倍的亮场下观察L929细胞形态并进行拍照。
1.4 数据处理与分析
每个实验均重复3 次,结果以平均值±标准差表示。用Excel软件进行数据处理,用Origin软件绘图。
2 结果与分析
2.1 酚基菊糖衍生物的合成
以菊糖为原料通过简单的三步反应合成酚类菊糖衍生物。首先,以NBS为溴代试剂与菊糖C6位伯羟基反应合成溴代菊糖(化合物1),因为在三苯基膦存在下NBS可以选择性地与伯羟基反应[11];然后,通过乙二胺与溴代菊糖的亲核反应将乙基氨基引入菊糖,制备6-N-(氨基乙基)-6-脱氧菊糖(化合物2);最后,羟基苯甲醛与化合物2末端基团氨基反应,在菊糖上引入具有C=N结构的酚基(化合物3)。
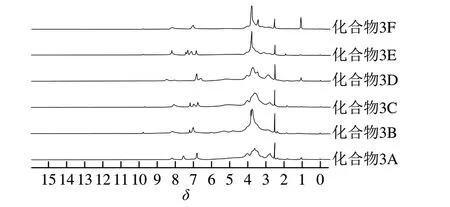
图3 菊糖衍生物的1H NMR图Fig. 3 1H NMR spectra of inulin derivatives
化合物3根据合成后R取代基的不同分为6 种,即化合物3A~F。对化合物2和化合物3A~F进行FTIR分析和1H NMR分析(图2、3)。

图2 菊糖及其衍生物的FTIR图Fig. 2 Fourier transform infrared spectra of intermediate products and inulin derivatives
化合物2:FTIR:3 353(-NH2和-OH)、1 648 cm-1(-NH2)。1H NMR(500 MHz,DMSO-d6):δ2.51(CH2-NH2)、2.62(CH2-NHCH2)和δ3.51、3.58(NH-CH2-CH2)。
化合物3A:FTIR:3 356(-NH2和-OH)、1 647 cm-1(N-H)和1 605、1 586 cm-1(苯环C=C)以及838 cm-1(苯环C-H)。1H NMR(500 MHz,DMSO-d6):δ2.77、2.86(CH2-NH-CH2),δ6.80、7.56(苯环-H)以及δ8.16(CH=N)。
化合物3B:FTIR:3 363(-NH2和-OH)、1 647(-NH-)、1 596 cm-1(苯环C=C)和873、790、693 cm-1(苯环C-H)。1H NMR(500 MHz,DMSO-d6):δ2.79、2.90(CH2-NH-CH2),δ7.01、7.2(苯环-H)和δ8.15(CH=N)。
化合物3C:FTIR:3 373(-NH2和-OH)、1 648(-NH-)、1 601(苯环C=C)、823 cm-1(苯环C-H)。1H NMR(500 MHz,DMSO-d6):δ2.78、2.85(CH2-NH-CH2),δ6.76~7.18(苯环-H),δ8.09(CH=N)。
化合物3D:FTIR:3 362(-NH2和-OH)、1 636 cm-1(-NH-)和784、737 cm-1(苯环C-H)。1H NMR(500 MHz,DMSO-d6):δ2.79、2.89(CH2-NH-CH2),δ6.57~6.82(苯环-H)和δ8.47(CH=N)。
化合物3E:FTIR:3 373(-NH2和-OH)、1 647(-NH-)、1 594 cm-1(苯环C=C)和877、827 cm-1(苯环C-H)。1H NMR(500 MHz,DMSO-d6):δ2.81、2.89、3.10(CH2-NH-CH2),δ3.67(OCH3),δ6.82~7.31(苯环-H)和δ8.19(CH=N)。
化合物3F:FTIR:3 385(-NH2和-OH)、1 644(-NH-)、1 605(苯环C=C)、835 cm-1(苯环C=H)。1H NMR(500 MHz,DMSO-d6):δ2.82、2.91(CH2-NH-CH2),δ3.44(OCH3),δ7.01(苯环-H)和δ8.15(CH=N)。
在FTIR图谱中,与菊糖相比,终产物菊糖衍生物3A~F在1 600 cm-1附近出现新的振动吸收峰,为苯环C=C键的伸缩振动峰,700~900 cm-1之间出现的新的吸收峰为苯环上C-H变形振动峰。这些峰都是苯环的特征吸收峰,由此表明酚基菊糖衍生物制备成功。在1H NMR谱图中,在δ3.0~5.0处的信号峰主要为糖骨架的氢峰,δ2.7~2.9之间的信号峰为亚胺及相连的亚甲基的氢峰,δ6.5~7.3之间为苯环上氢质子的信号峰;δ8.1附近的信号峰为C=N上的氢峰。δ2.7~2.9、6.5~7.3和δ8.1附近的3 组峰均为苯环及C=N的特征峰,进一步证明酚基成功连接到菊糖上。
2.2 酚基菊糖衍生物抗氧化活性
抗氧化剂的抗氧化活性可以通过多种途径实现,包括清除分子氧或降低分子氧局部浓度、去除金属离子、捕获O·或过氧化氢等活性氧(reactive oxygen species,ROS),清除·OH、RO·或ROO·等可以引发链反应的自由基,猝灭氧单质[27]。为研究这些酚基菊糖衍生物的抗氧化活性,测定其自由基清除能力、还原能力和Fe2+螯合能力。
2.2.1 自由基清除能力
自由基是一类含有一个或多个未配对电子的活性物质[12,28]。体内的自由基大多是ROS或活性氮。本实验选择2 种典型的ROS(O·和·OH)和一种活性氮(DPPH自由基),考察酚类菊糖衍生物的自由基清除能力。
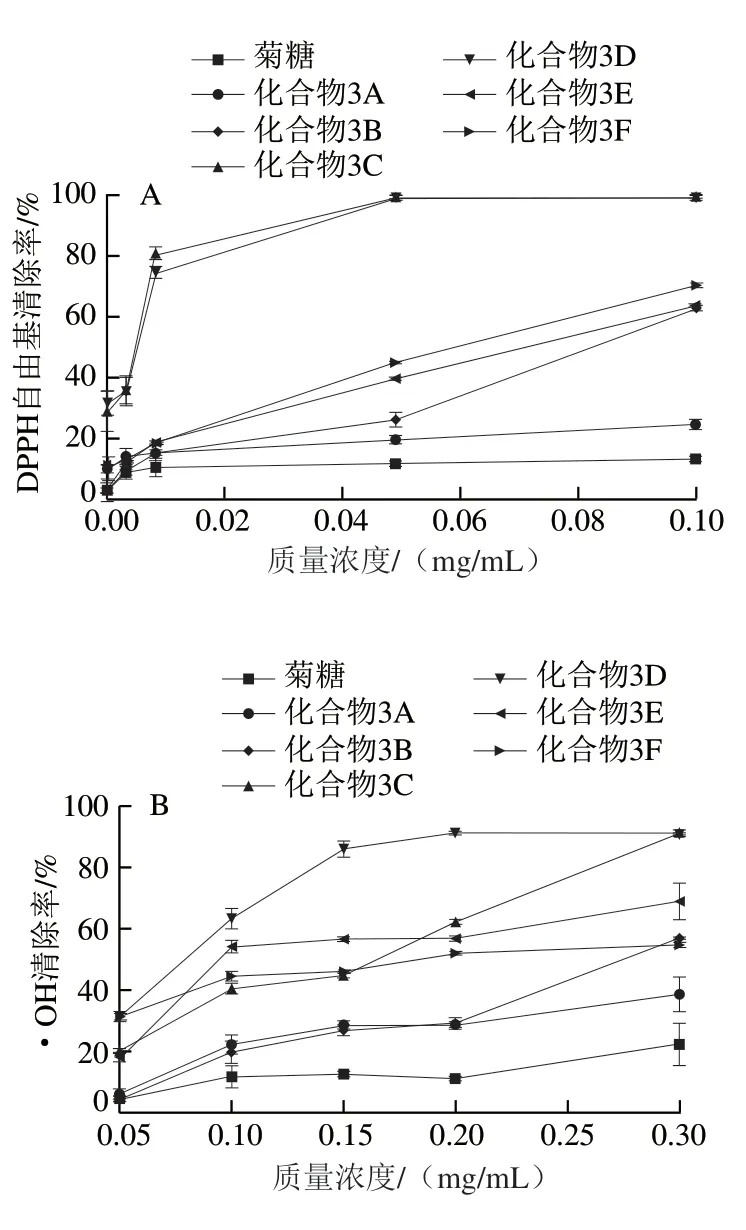
由图4可知,菊糖与菊糖衍生物对DPPH自由基的清除能力均随质量浓度的增大而增大。菊糖本身的DPPH自由基清除能力有限,在实验质量浓度范围内对DPPH自由基清除率最高仅达到13.9%(0.1 mg/mL),对·OH清除率最高只有20.68%(0.3 mg/mL),对O2-·清除率最高只有22.4%(0.2 mg/mL)。与菊糖相比,菊糖衍生物具有更强的自由基清除能力,说明酚基的存在增强了菊糖衍生物的自由基清除能力。此外,连接双酚的菊糖衍生物(化合物3C、3D)较连接单酚的菊糖衍生物(化合物3A、3B和化合物3E、3F)的自由基清除能力高。连接双酚的化合物3C、3D对DPPH清除能力的IC50均为0.007 mg/mL,较VE(IC50为0.01 mg/mL[29])的DPPH自由基清除能力高;化合物3C清除O2-·的IC50为0.042 mg/mL。
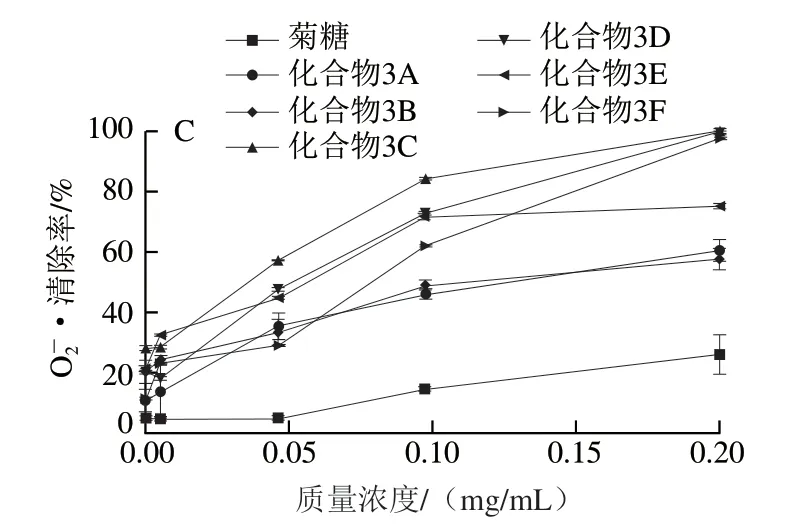
图4 不同质量浓度菊糖和菊糖衍生物的DPPH自由基(A)、·OH(B)和O2-·(C)清除能力Fig. 4 DPPH radical (A), ·OH (B) and O2-· (C) scavenging capacity of inulin and inulin derivatives at different concentrations
酚类化合物在清除自由基的过程中,一般是通过失去H原子形成芳氧自由基(中间体1、2),H原子在苯环上相邻的两个氧之间快速交换,共振体的存在降低了体系内能,使芳氧自由基更稳定。形成的自由基越稳定,该化合物的自由基清除能力越强。本实验制备的酚类菊糖衍生物中,C=N与苯环形成共轭结构,有助于形成稳定的芳氧自由基(图5),相应的酚类菊糖衍生物的清除自由基能力就更强。有文献报道,邻苯二酚是多酚类化合物提供电子或H原子的关键决定因素[30]。

图5 菊糖衍生物清除自由基的可能机理Fig. 5 Possible free radical scavenging mechanism for inulin derivatives
2.2.2 Fe2+螯合能力
过渡金属离子可以引发自由基反应并引起脂质过氧化和心血管疾病,造成DNA损伤[31]。螯合剂可以降低氧化还原电位,能作为辅助抗氧化剂控制金属离子的氧化,并稳定金属离子的氧化形式。因此,对过渡金属离子的螯合能力是与自由基清除能力密切相关的一种抗氧化能力。从图6可以看出,在实验质量浓度(0.001~0.15 mg/mL)范围内,菊糖的Fe2+螯合能力不强,当质量浓度为0.15 mg/mL时,菊糖的Fe2+螯合率为14.1%。连接酚基后,菊糖衍生物的Fe2+螯合能力明显提高,化合物3B的 Fe2+螯合能力的IC50能够达到0.006 mg/mL。
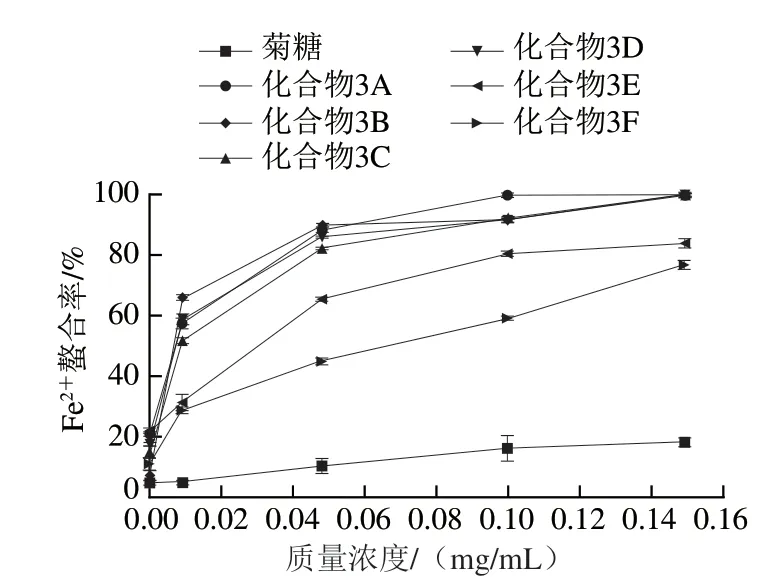
图6 不同质量浓度菊糖和菊糖衍生物的Fe2+螯合能力Fig. 6 Fe2+ chelating ability of inulin and inulin derivatives at different concentrations
2.2.3 还原能力
化合物的还原能力可以作为评价其潜在抗氧化活性的重要指标[32]。由图7可知,菊糖本身的还原能力很弱,连接酚基之后还原能力有所提高。整体来看,制备的酚基菊糖衍生物中,化合物3A、3B、3E、3F的还原能力较为接近,化合物3C、3D的还原能力明显较强。这与自由基清除能力的结果相似,邻二双酚结构的存在使得酚基菊糖衍生物具有更高的还原能力。一方面,邻位的羟基通过共轭效应供电子使得酚羟基上的氧电负性增大,还原能力增强;另一方面,分子中活性羟基的数量也是影响其还原能力的因素。因此,化合物3C和3D的还原能力更强。
图7 不同质量浓度菊糖和菊糖衍生物的还原能力Fig. 7 Reducing power of inulin and inulin derivatives at different concentrations
2.3 菊糖及其衍生物的生物相容性
以小鼠成纤维细胞L929为模型,通过CCK-8法测定菊糖及其衍生物的细胞毒性,以评价其生物相容性。从图8可以看出,在0~125 μg/mL的质量浓度范围内,没有观察到明显的细胞毒性,但更高的质量浓度(250 μg/mL)会导致细胞存活率轻微下降(下降约10%)。L929细胞形态一般呈特征性纺锤形(图9A~D),在质量浓度为250 μg/mL时,细胞形态部分改变,可以发现少量溶出物(图9E),进一步证明了细胞毒性实验结果。这些结果表明,在质量浓度低于125 μg/mL时,带有酚基的菊糖衍生物对L929细胞具有良好的生物相容性。这意味着这些菊糖衍生物在不损伤正常细胞的情况下具有较好的抗氧化活性。有研究[33-35]表明,经化学改性制备的酚类壳聚糖衍生物可以作为具有抗氧化功能的食品添加剂及食品包装材料。本实验制备的这些菊糖衍生物在具有优良抗氧化活性的同时也有较好的生物相容性,这些实验数据为酚类菊糖衍生物在食品中的应用奠定了一定的基础。后期会继续展开体内抗氧化实验及体内毒性实验进一步确定酚类菊糖衍生物的食品安全性。
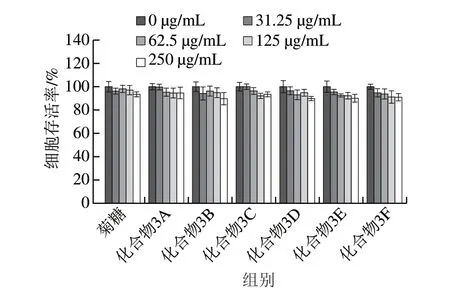
图8 菊糖和菊糖衍生物的L929细胞毒性Fig. 8 Cytotoxicity of inulin and inulin derivatives to L929 cells
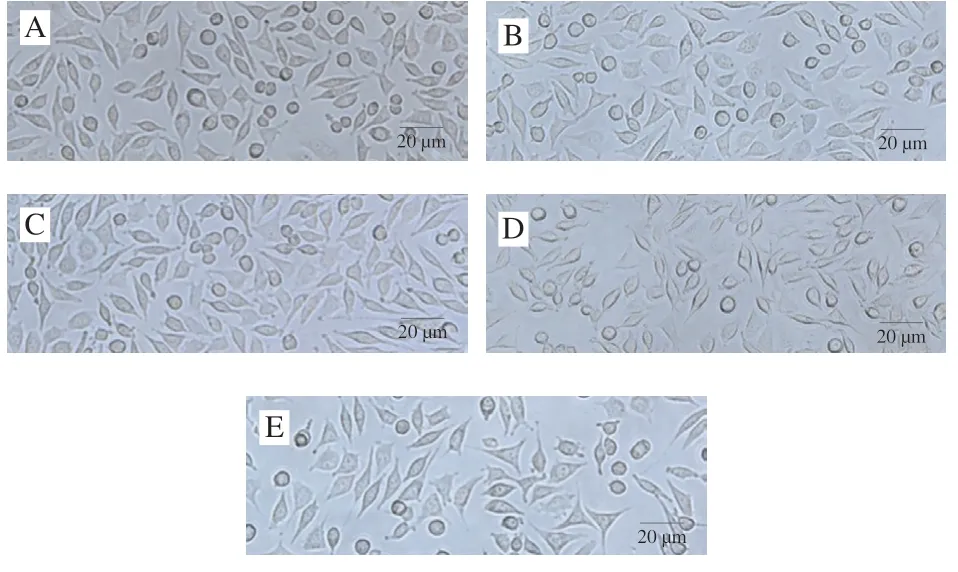
图9 经菊糖衍生物化合物3C处理24 h后L929细胞的生长情况Fig. 9 Microscopic images of inulin derivative 3C-treated L929 cells for 24 h
3 结 论
本实验通过化学修饰在菊糖分子上引入具有高抗氧化活性的酚类基团,合成了6 种酚类菊糖衍生物。通过测定自由基清除能力、还原能力和Fe2+螯合能力等,研究酚类基团对菊糖抗氧化活性的影响。结果表明,酚基的引入极大地提高了菊糖衍生物的抗氧化活性,且体外细胞实验结果表明菊糖衍生物具有良好的生物相容性,有望应用于食品和保健品领域。
猜你喜欢
中学化学(2022年5期)2022-06-17
华南师范大学学报(自然科学版)(2021年4期)2021-08-30
陶瓷学报(2020年6期)2021-01-26
当代水产(2020年4期)2020-06-16
当代水产(2020年3期)2020-06-15
理科考试研究·高中(2019年8期)2019-09-19
世界农药(2019年3期)2019-09-10
科学中国人(2018年8期)2018-07-23
中学生数理化·高二版(2016年3期)2016-12-26
化学教学(2015年11期)2015-12-19
