
10%噁嗪草酮悬浮剂高效液相色谱串联质谱分析
2021-12-15 03:19石先罗卞传飞董泽民李保同
应用化工 2021年11期
石先罗,卞传飞,董泽民,李保同
(1.江西农业大学 国土资源与环境学院,江西 南昌 330045;2.江西水利职业学院 水利工程系,江西 南昌 330013)
噁嗪草酮(Oxaziclomefone),分子式为C20H19Cl2NO2,结构式见图1[1]。该制剂是一种内吸传导性的有机杂环类除草剂,主要适用于水稻稻田杂草的防除,可用于秧田、直播田及移栽田[2-3]。目前关于噁嗪草酮原药和残留检测方法主要有气相色谱法、高效液相色谱法等[4-5]。气相色谱法由于噁嗪草酮具有沸点高,难以气化,需要对样品进行前处理费时费力。高效液相色谱法检测30%噁嗪草酮悬浮剂则保留时间较长(15.3 min),不利于快速开展[6]。本文在上述研究基础上,建立了高效液相色谱串联质谱检测10%噁嗪草酮有效成分分析方法,该方法操作简便,可用于检测分析10%噁嗪草酮悬浮剂。
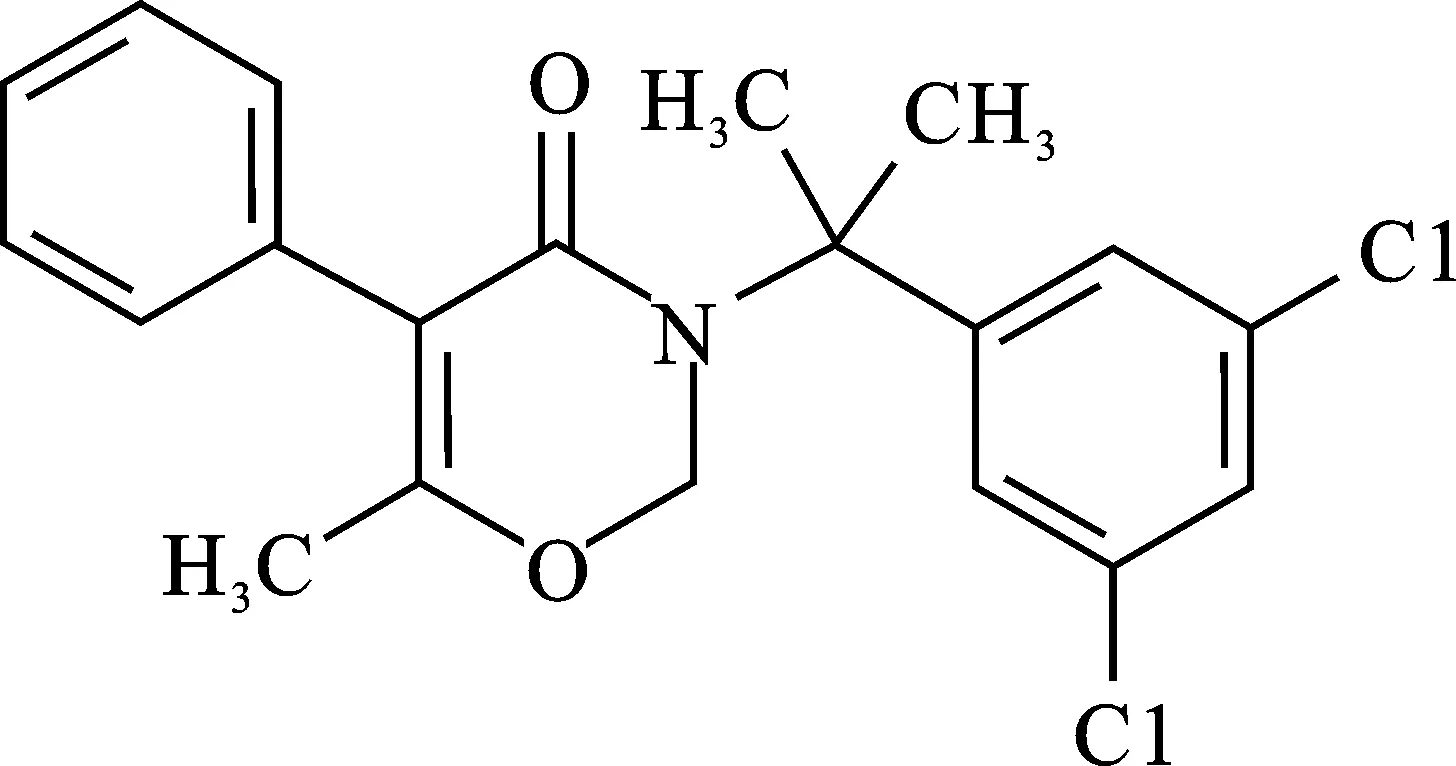
图1 噁嗪草酮化学结构式Fig.1 Oxaziclomefone chemical structural formula
1 实验部分
1.1 材料与仪器
乙腈、甲酸(88%)均为色谱纯;超纯水;10 mg噁嗪草酮标样(纯度99.3%),由北京曼哈格生物科技有限公司提供;10%噁嗪草酮悬浮剂,由山东先达农化股份有限公司提供。
Agilent-1260的高效液相色谱仪(HPLC)含配套全自动进样器,Zorbax Eclipse XDB-C18(4.6 mm×150 mm,5 μm)色谱柱;Agilent-6120质谱仪(MS,含单级四级杆液质联用仪)。
1.2 实验方法
1.2.1 标准溶液的配制 取噁嗪草酮标准样品 10 mg(纯度99.3%)溶于100 mL容量瓶,加入乙腈超声波溶解、定容、摇匀,此溶液为100 mg/L噁嗪草酮储备液[7]。用乙腈按梯度依次稀释上述噁嗪草酮储备液,至6个浓度梯度依次为4.0,1.0,0.5,0.1,0.01,0.001 mg/L。
1.2.2 试样溶液的配制 准确称取10%噁嗪草酮悬浮剂200 mg(精确至1 mg),转移至50 mL容量瓶,加入乙腈作为溶剂,置于实验室超声波仪助溶 5 min 以上直至完全溶解,后定容、摇匀,配制成 400 mg/L。取0.5 mL上述配制溶液置于50 mL容量瓶,加入乙腈定容,得到4 mg/L的噁嗪草酮试样溶液,冷藏备用。
1.2.3 测定 将配制好的溶液过0.22 μm有机滤膜,在上述设定好的仪器条件下,待基线平稳后,按照进样顺序连续进样,依次为标样→试样→试样→标样,得到10%噁嗪草酮色谱图见图2。
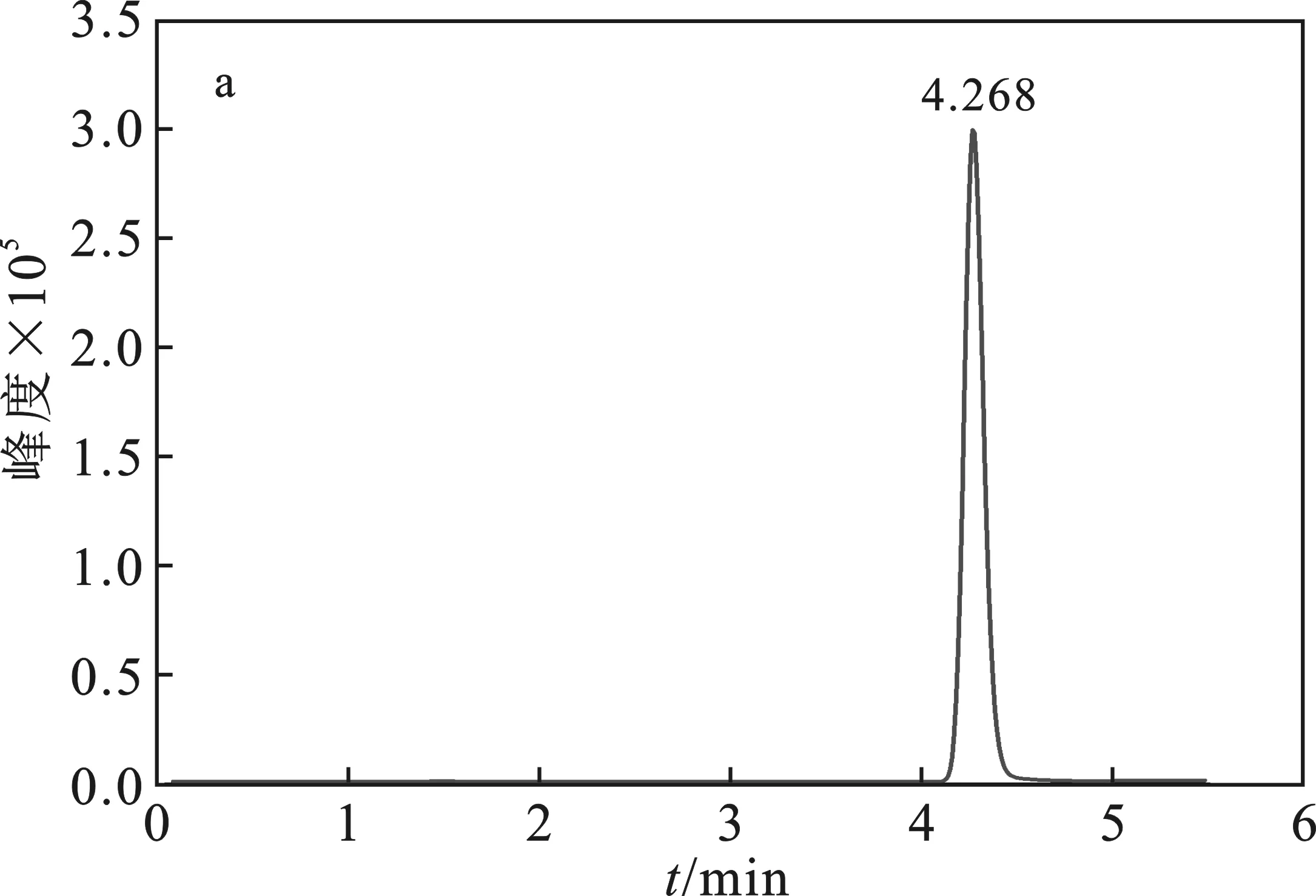
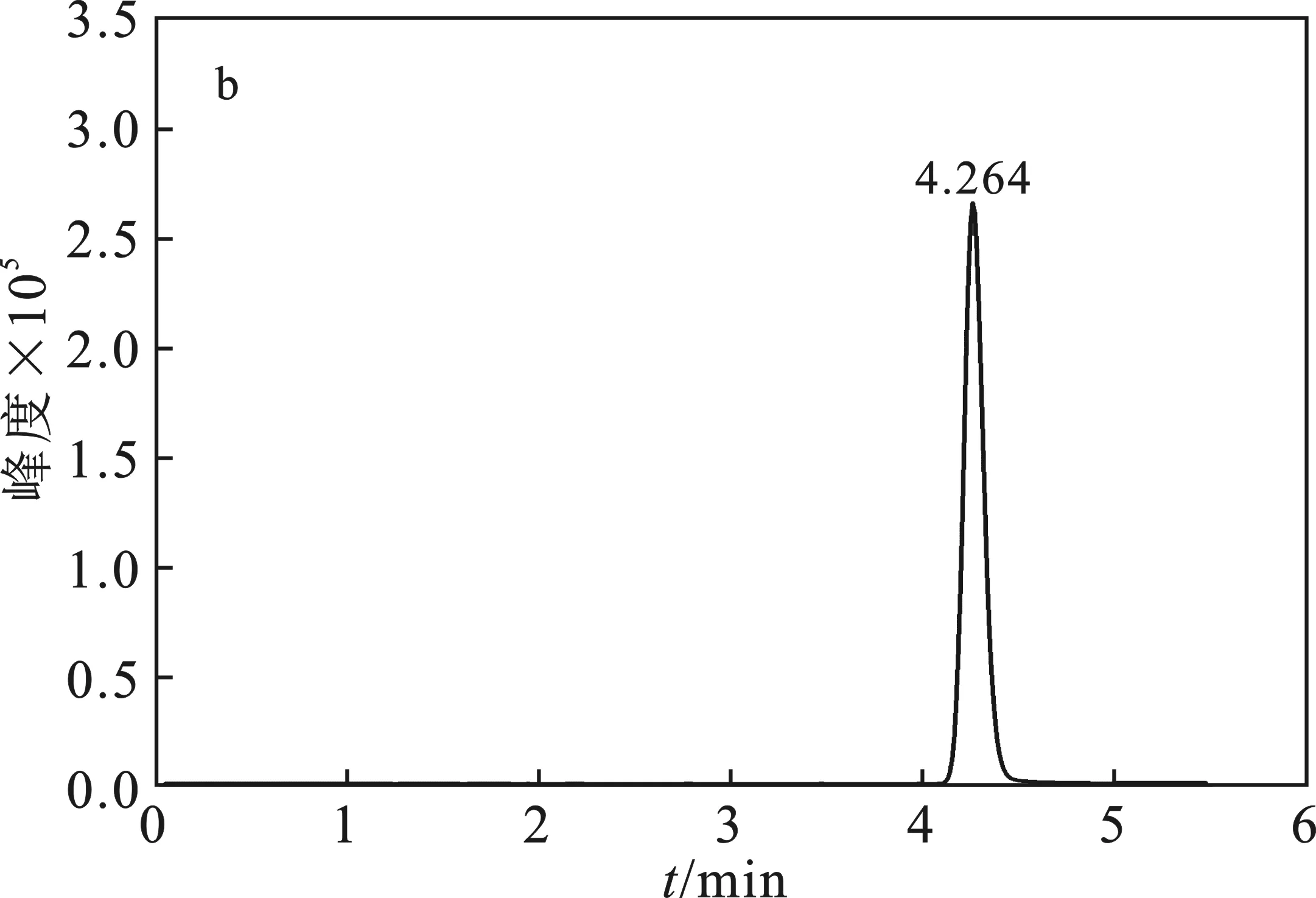
图2 噁嗪草酮色谱图Fig.2 Chromatogram of oxaziclomefonea.10%噁嗪草酮悬浮剂;b.标样,质量浓度均为1 mg/L
1.2.4 计算 利用上述测得的标准样品溶液和试样溶液中噁嗪草酮峰面积,根据如下公式计算试样溶液中噁嗪草酮质量分数(w2,%):
(1)
式中A1——标准样品中噁嗪草酮峰面积;
A2——试样中噁嗪草酮峰面积;
M1——噁嗪草酮标准样品质量,g;
M2——噁嗪草酮试样质量,g;
w1——取99.3%为标准样品质量分数,%。
2 结果与讨论
2.1 色谱条件的确定
为找出最佳峰型及最佳保留时间,利用1 mg/L噁嗪草酮标准溶液对色谱条件进行优化,根据文献研究发现乙腈-水流动相能较好地分离出峰型,但是检测过程中发现较多杂质峰干扰,目前主要是以添加磷酸调整pH值为主。本次实验中发现采用 0.1% 的甲酸相对于0.1%磷酸能获得更好的峰型且主峰与杂峰完全分离,较传统方法提高了峰的对称性及提高了反应灵敏度。
同时为了获得更好的峰形,同时减少杂质干扰,对乙腈与0.1%甲酸的体积比进行了多次调整,结果见表1。

表1 不同体积比的乙腈-0.1%甲酸水溶液对噁嗪草酮标样保留时间和峰面积的关系Table 1 Relationship between retention time andpeak area in acetonitrile-formic acid aqueous
由表1可知,当乙腈-0.1%甲酸体积比为60∶40时噁嗪草酮保留时间为15.715 min,当比例为 70∶30 时噁嗪草酮保留时间为7.714 min;当体积比为80∶20时,时间为4.226 min,此时峰形更好,保留时间适中,相对于已有方法提升了检测效率;当体积比为90∶10时,噁嗪草酮保留时间2.681 min,虽然缩短了时间但是与杂质峰时间接近。而后调整流动相的流速,相比0.8 mL/min,在1.0 mL/min的流速下其峰型更好,且多次进样的结果更稳定,由此确定最终色谱条件[8]。最终色谱条件确定为:进量设置为5.00 μL,V乙腈∶V甲酸=80∶20,柱温设置为 35 ℃,流速设置为1 mL/min。
2.2 质谱条件的确定
选择scan,采用ESI离子源(+/-)对1 mg/L噁嗪草酮标准溶液全扫描,在正离子模式下可以获得最强有效峰,因此确定正离子模式进行扫描[9-10]。由于噁嗪草酮的相对分子质量为376.3,所以在相对分子质量为200~500的范围内进行全扫描,结果表明:在m/z为376.1时为最强有效峰。在SIM模式下,对碰撞诱导解离电压等参数优化调整,使得仪器响应值最大化,由此确定最终质谱条件。最终质谱条件为:SIM质荷比选择376.1(m/z),介质气体选择干燥氮气,温度设置为350 ℃,流速设定为 12.0 L/min,碰撞诱导解离为100 V,毛细管电压选择正负3 000 V,雾化气压力35.0 kPa,驻留时间 590 ms,相对驻留选择100%。
2.3 分析方法的线性相关性
制备质量浓度梯度为0.001,0.01,0.1,0.5,1,4 mg/L的噁嗪草酮试样溶液,根据上述确定的液质联用检测条件,将噁嗪草酮质量浓度设为x轴,检测得到的峰面积为y轴,线性回归方程:y=2 168 083x+8 094.45(R2=0.999 9)。结果表明,其线性关系良好,线性关系见图3。根据国家标准,实际进样时仪器的信噪比(S/N)为3时,为该物质的最低检出限,得出该仪器条件下噁嗪草酮的最低检测限为 0.001 mg/L。

图3 噁嗪草酮线性关系图Fig.3 Linear relationship of oxaziclomefone
2.4 分析方法的精确度
从10%噁嗪草酮试样溶液中准确吸取5份样品,待仪器基线平稳后,在上述液质条件下连续进行测定[11]。计算得出测定10%噁嗪草酮悬浮剂的结果标准偏差和变异系数分别是0.087,0.81%(见表2),符合CIPAC的要求(<1.17%),证明此方法的精密度符合要求。
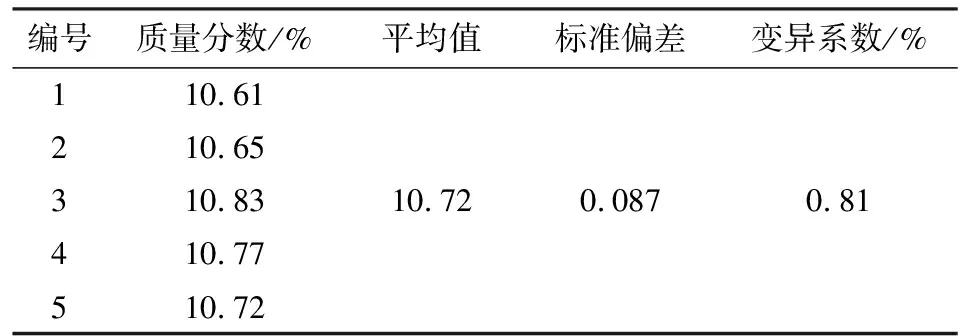
表2 10%噁嗪草酮悬浮剂精密度测定Table 2 Precision of oxaziclomefone 10% SC
2.5 分析方法的准确度
分别将5份等质量的10%噁嗪草酮悬浮样品移于100 mL容量瓶中,并分别加入一定量的噁嗪草酮标准溶液,在液质条件下进行定量检测[12]。结果表明(见表3),该条件下噁嗪草酮的回收率在 98.235%~100.57%之间。

表3 10%噁嗪草酮悬浮剂准确度测定Table 3 Accuracy determination ofoxaziclomefone 10% SC
3 结论
(1)在色谱优化过程中,分别采用了乙腈-磷酸水和乙腈-甲酸水,结果发现乙腈-0.1%甲酸水具有更好效果。对比例进行优化,乙腈和甲酸配比分别采用了60∶40,70∶30,80∶20,90∶10的比例,结果表明,体积比为80∶20、温度为35 ℃时噁嗪草酮保留时间适中、峰形最优且能与杂质具有较好的分离,因此采用该比例作为流动相。
(2)噁嗪草酮检测结果表明,利用高效液相色谱串联质谱测定10%噁嗪草酮悬浮剂含量检测结果准确可靠、操作快速便捷,可用于10%噁嗪草酮悬浮剂的快速定量检测分析。
猜你喜欢
煤化工(2022年3期)2022-07-08
色谱(2021年7期)2021-06-07
农药科学与管理(2019年8期)2019-11-23
农药科学与管理(2019年5期)2019-08-13
农药科学与管理(2019年10期)2019-04-20
中国蜂业(2018年4期)2018-05-09
当代化工研究(2016年6期)2016-03-20
中国资源综合利用(2016年10期)2016-01-22
西北园艺(果树)(2015年1期)2015-02-21
无机化学学报(2014年3期)2014-02-28
