
中药一致性评价关键技术
——定量指纹图谱联合 一线多评法研究
2021-12-30 03:13闫慧孙国祥孙万阳兰丽丽李想蒲道俊胡延雷贾景明陈振鸿沈阳药科大学中药学院沈阳00沈阳药科大学药学院沈阳00暨南大学药学院广州03西南药业股份有限公司重庆00038国药集团工业有限公司北京030新疆富沃药业有限公司新疆阿克苏地区899
中南药学 2021年11期
闫慧,孙国祥,孙万阳,兰丽丽,李想,蒲道俊,胡延雷,贾景明,陈振鸿(.沈阳药科大学中药学院,沈阳 00;.沈阳药科大学药学院,沈阳 00;3.暨南大学药学院,广州 03;.西南药业股份有限公司,重庆 00038;.国药集团工业有限公司,北京 030;.新疆富沃药业有限公司,新疆 阿克苏地区 899)
药物一致性评价,狭义上要求仿制药与原研药符合相同的质量标准,广义上要求两者具有相同的活性成分和适应证,药物剂型规格和给药途径相同,符合相同的质量标准,达到生物等效,即药学等效和药效等效[1]。由于中药(traditional Chinese medicine,TCM)没有进行过系统的标准化过程,因此无法采用参比制剂来进行一致性评价。与化药相比,中药来源复杂,药效成分多样,对某种疾病起效可能为几种成分的协同作用,故仅检测其中的一种或几种成分不能达到整体评价中药质量的目的,这给中药的质量评价带来很大的困难。因此,为了更加系统准确地评价中药的质量,孙国祥教授建立了中药定量指纹图谱评价理论和技术,提出中药标准制剂控制模式和全质量关控制模式。中药标准制剂控制模式是中药一致性评价首选模式,尤其是双标校正后的标准制剂的对照指纹图谱将是中药一致性评价的必由之路。中药系统指纹定量法从宏观定性和宏观定量角度出发,以定量指纹图谱控制中药中整体化学物质,进而成为整体评价中药质量的新方法[2]。本文在中药标准制剂建立后,采用定量指纹图谱技术模式即中药指纹图谱与多指标成分定量分析相结合的中药质量控制模式[3]。
但由于中药成分复杂,某些对照品价格昂贵或难得,使得某些成分的定量控制比较困难,因此,王智民等[4]提出了一测多评法。一测多评法的出现不仅减少了中药质量评价的成本,也简化了测定过程,提高了工作效率。一测多评法是通过中药有效成分间存在的内在函数关系和比例关系,建立样品中某一有效、价廉、易得的典型成分与样品其余成分间的相对校正因子(relative correction factor,RCF)以计算样品中其他成分的量[4]。但是一测多评法在技术上存在4个方面的不足:① 缺乏线性范围考察和系统方法学验证技术,导致误差较大;② 无误差分析理论和可靠度分析理论,导致分析方法可靠度无法验证,也无法指导试验技术人员如何避免误差;③ 色谱体系无误差校正方法,通常检测色谱条件与当初建立标准的色谱系统存在系统定量误差,误差传递导致最终结果误差较大(大于5%);④ 理论技术不够扎实,数学上理论探讨不够深入。
本文在一测多评法的基础上,提出一线多评法(multi-markers assay by monolinear method,MAML),即基于一测多评法原理并结合中药定量指纹图谱理论提出的新方法,弥补了一测多评法在技术上的缺陷和不足。既然中药定量指纹图谱承认所确定的全部指纹峰都能整体定量,那么对于主要的几个指标成分的量值分布必然有确定的函数关系,因此必然能使用一测多评法。一测多评法最大缺陷是忽略了线性范围,考察浓度范围太窄,忽视了建立标准系统和检验系统之间的量值校正问题即系统定量误差没进行任何校正。一线多评法是在多指标定量时采用与指纹图谱检测相同的色谱条件同时建立多个活性化合物的标准曲线,利用一测化合物(一测物)和多评化合物(多评物)标准曲线的参数计算校正因子、误差、可靠度,从而实现用一测物标准曲线对多评物进行定量的分析方法。当方法建立后,以一线多评法对多评物进行定量时的计算公式与一测多评法相同(一般以线性范围的平均浓度进行试验),不同的是相对校正因子由多条标准曲线的参数计算而来,具有误差分析和校正理论及可靠度分析理论。因此一线多评法是在定量指纹图谱基础上利用多条标准曲线获得的与定量指纹系统具有密切关联度量值的相对校正因子法,由于有双标校正法而实现定量误差可控或很小。中药定量指纹图谱联合一线多评能实现对中药化学指纹物质总量的等位等效控制。 孙国祥教授提出:① 中药多指标定量分析属于中药质量的多点或者多点线质量控制模式(两点确定一直线,因此是线性控制范围);② 定量指纹图谱控制属于从中药主组分化学成分出发形成的平面轮廓控制(用指纹图谱定性控制是形象化外观圆控制,用中药指纹图谱定量控制是对总量的幅度控制-大小不同圆);③ 中药溶出度控制属于对中药主组分指纹溶出的立体控制-球形(实际情况可能为凹凸不规则的球)。中药质量一致性评价必须是基于标准制剂的点线面立体化控制,见图1。中药质量一致性评价的药学质量研究应从这三个方面开展,才能控制好药效物质总量的等位,同时控制好体外溶出度一致性来实现对中药固体制剂工艺过程的一致性控制——质量与药效实现等位等效。
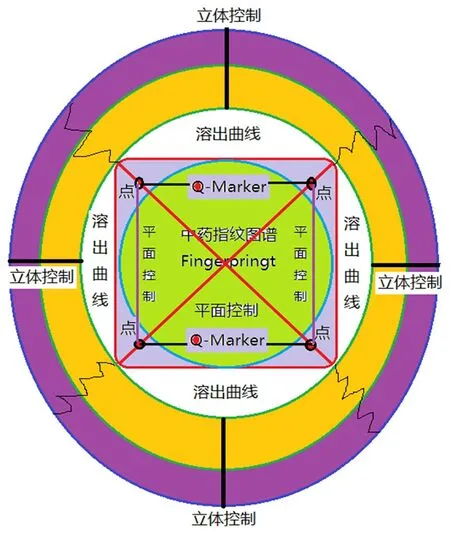
图1 中药一致性评价点线面立体控制模型(Q-markers点线控制,指纹图谱圆面控制和溶出度立体球控制)Fig 1 Dot-line-plane-stereoscopic control model for TCM consistency evaluation(dot-line control of Q-markers,circular plane control of fingerprints and stereoscopic ball control of dissolution)
1 定量指纹图谱联合一线多评法模型的建立
1.1 一线多评法线性范围
在定量指纹图谱条件下的多指标定量时,必须采用统一化色谱条件同时建立多条标准曲线,用其中一条标准曲线来对多指标分别准确定量的方法称为一线多评法。一线多评法的最大特点是把标准曲线法最大程度地简化,节省对照品使用。具体要求为:① RCF(fsi)必须满足一定线性范围,推荐10~20倍浓度范围,最大不超过100倍;② 一测物线性范围为Rs=[Cs1,Cs2],多评物线性范围为Ri=[Ci1,Ci2],则被测物浓度应在Ri=[Ci1,Ci2],通常线性范围的均值浓度为最佳点Cavg=(C1+C2)/2;③ 用标准曲线A=bC+a求RCF更合理(有线性范围)。用线性方程求RCF特点:① 用最小二乘法计算浓度斜率bi,能准确地揭示峰面积对浓度增长的灵敏度,且给出误差项ai(标准曲线的截距);② 给出的线性范围和相关系数能揭示浓度适用范围和误差大小;③ 用截距和截距斜率容易计算fsi误差;④ 方法可靠度能预先准确估算;⑤ 一线多评法方法学验证后,仍然采用一点法或两点法对多指标成分定量,因此检验工作中没必要每次都作标准曲线,避免浪费对照品和资源。
1.2 一线多评法应用
一测物S标准曲线和多评物i标准曲线见公式(1)和(2),根据标准曲线计算一测物绝对校正因子fi见公式(3)和(4)。bas=as/Cs,bai=ai/Ci分别为一测物S截距斜率和多评物i截距斜率(即斜率绝对误差)。可知测定绝对校正因子时,一测物Cs和多评物Ci对bas和bai影响很大,两者浓度越大则截距斜率越小,即高浓度测定误差很小。线性平均浓度C0=0.5(C1+C2)(即通常为测定样品浓度,线性范围中间浓度)为最佳。用斜率公式直接计算相对校正因子fsi见公式(5),把标准曲线斜率之比称为简相对校正因子f'si,当ba=│a/C│≤1%b时可用斜率直接计算fsi,见公式(6)(误差小于1.0%)。

一线多评法是建立在一测物S和多评物i的标准曲线基础上,具有以下特点:① 线性范围揭示一线多评法适用范围;② 给出误差范围和可靠度;③ 基于标准曲线的一线多评法科学性和准确性显著提高;④ 一线多评法只有在精密度和方法重复性均ba=│a/C│≤1%b时,RSD≤2.0%才能保证方法的准确度是可靠的。
1.3 一线多评法误差
1.3.1 相对校正因子的相对误差 按误差传递规律得fsi相对误差见公式(7),主要由一测物S和多评物i截距误差之差所决定。用均值浓度计算截距斜率时,标准曲线自身误差的大小决定了相对fsi误差。计算一线多评的被测物浓度的相对误差,见公式(8),称量步骤的相对误差小是十分重要的,得定量可靠度,见公式(9)。其中被测物相对峰面积误差代入样品中i组分精密度的RSDi(REi≈RSDi),一测物S的相对峰面积误差代入对照品精密度试验中的RSDs(REs≈RSDs),两者有部分抵消作用(之差小于1.0%):
① 若△fsi/fsi≤2.0%和△Csi/Cs≤2.0%,则定量误差小于4.0%;定量可靠度R不低于95.5%;
② 若△fsi/fsi≤1.0%和△Csi/Cs≤1.0%,则定量误差小于2.0%;定量可靠度R不低于97.7%;
③ 截距斜率误差大小直接影响定量结果,一测物和多评物浓度越高则截距斜率越小,测定样品准确度越好。
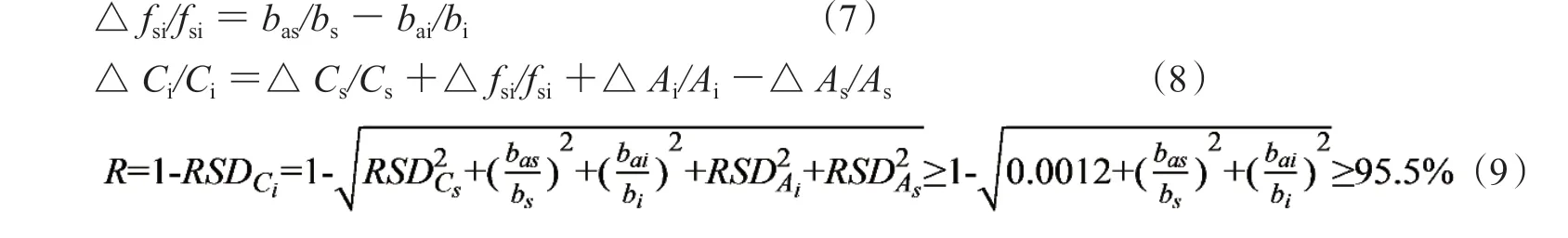
1.3.2 一线多评法的相对误差 如果标准曲线仅有截距误差(制备标准溶液的浓度误差小于1.0%),则测定多评物的可靠度,见公式(9),① 测定S和i峰面积均RSD≤2.0%(不含中间精密度),截距误差小于1.0%,则方法可靠度大于96.7%;② 若五项相对误差都小于1.0%,则方法误差小于2.0%,可靠度大于97.7%,称量误差小显得十分重要;③ 若五项相对误差都小于2.0%,则方法误差小于4.0%,可靠度大于95.5%;④ 当斜率误差较大时方法准确度误差约5.0%。当只考虑截距误差时,可靠度估算见表1。因为一线多评法的相对误差只与测定混合对照品和多组分样品时的仪器精密度试验结果和称量精密度有关。因此一线多评法的方法重复性试验和准确度试验是评价该方法整体误差的必要方法。根据各测量误差进行不确定度和可靠度大小进行标准划分,总计有9个等级,见表1。
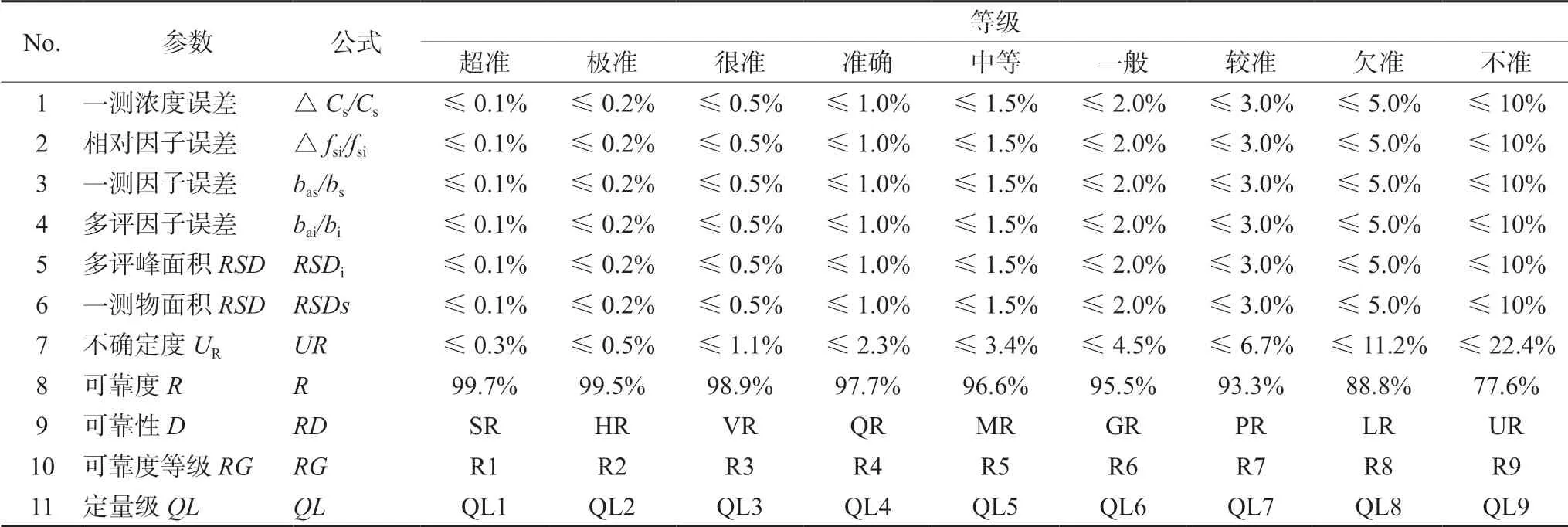
表1 一线多评法的不确定度和可靠度划分标准 Tab 1 Unreliability and reliability of multi-markers assay by monolinear method
1.3.3 系统指纹定量法原理[5-8]系统指纹定量法(systematically quantified fingerprint method,SQFM)能从宏观定性角度和宏观定量角度对中药进行全方位立体量化控制,适合中药质量一致性评价。主要包括三个参数:宏定性相似度(Sm),宏定量相似度(Pm)和指纹均化变动系数(α),分别见公式(10~12)。其中α在分级控制时使用较少,只要充分利用好Sm和Pm就能实现对整体主组分指纹的定量控制,该方法还可用于紫外全指纹溶出度评价,同时还能以标准制剂(或参比制剂)为参照对不同批次中药固体制剂溶出曲线的相似性进行评价,因此SQFM和中药溶出系统指纹定量法是中药一致性评价的核心方法,它们的计算方法相同。
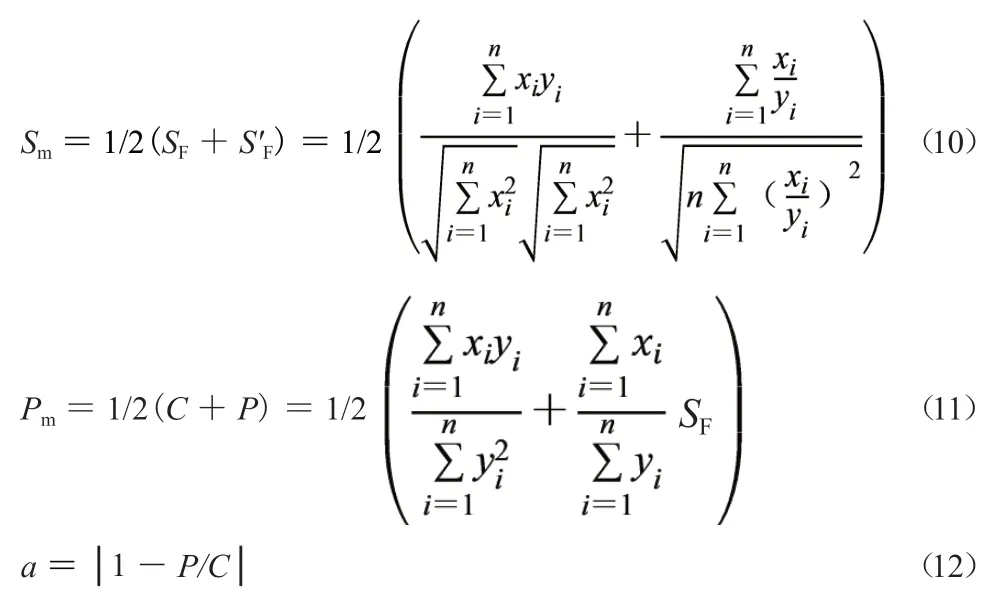
1.3.4 定量指纹图谱双标校正法 色谱系统改变会带来定量误差,通过双标校正法进行校正。把建立特征指纹图谱(标准指纹图谱)的系统称为第一色谱系统(first chromatographic system,FCS),对发生误差后的色谱系统称为第二色谱系统(second chromatographic system,SCS),在 强 极 性 区 和弱极性区各选择一个参照物峰作双标(一般为Q-markers),测定固定浓度双标混合溶液对应峰面积来计算FCS和SCS的绝对定量校正因子fd1和fdi,见公式(13)和公式(14)[9]。SCS与FCS的绝对定量校正因子之比称为双标相对定量校正因子fqi,见公式(15),把FCS定量性质平移到新检测系统可通过相对定量校正因子fqi与样品称样质量(mi)直接相乘实现校正,见公式(16),称为双标校正法[9]。双标校正法能消除不同色谱系统在不同时间测定的指纹图谱时所产生的系统误差,也能保证一线多评法误差可控或很小。双标校正法是定量指纹图谱执行中药质量一致性评价的必要基础和有效保证,是一线多评法应用的前提条件。
2 方法
2.1 仪器与试药

Agilent1100 型液相色谱仪(配有二极管阵列检测器、四元低压梯度泵、在线脱气装置、自动进样器),Agilent OpenLAB CDS Chemstation(Edition C.01.07)网络工作站(Agilent科技有限公司)。Sarturius-BS 110S分析天平(北京赛多利斯天平有限公司);ES-E120D电子分析天平(天津市德安特传感技术有限公司);超声波清洗机(深圳市洁盟清洗设备有限公司);安捷伦706ds全自动溶出仪(配有850ds自动取样器)。
磷酸(色谱纯,成都市科龙化工试剂厂);甲醇、乙腈(色谱纯,山东禹王和天下新材料有限公司);娃哈哈纯净水(沈阳娃哈哈启力食品有限公司);庚烷磺酸钠(色谱纯,山东省禹城市中美色谱产品厂);吗啡(MP,批号:171201-200822)、磷酸可待因(MMP,批号:171203-200504)、甘草苷(LQN,批号:111610-200604)、甘草酸铵(CHAA,批号:110731-201619)和苯甲酸钠(SB,批号:100433-200301)(对照品,中国食品药品检定研究院)。
复方甘草片(S1~S49为厂家A生产;S50~S61为厂家B生产;S62~S73为厂家C生产;S74~S85为厂家D生产;S86~S97为 厂 家E生 产;S98~S109为 厂 家F生 产;S110~S121为厂家G生产;S122~S133为厂家H生产;S134~S145为厂家I生产)。
2.2 溶液的制备
2.2.1 双标溶液制备 分别取MP和CHAA对照品适量,精密称定,加甲醇制成每1 mL含200 μg MP和800 μg CHAA的双标混合对照品溶液,摇匀,即得。
2.2.2 混合对照品溶液制备 分别取MP、LQN、MMP、SB和CHAA对照品适量,精密称定,加甲醇制成每1 mL含35 μg MP、60 μg LQN、16 μg MMP、160 μg SB和800 μg CHAA的混合对照品溶液,摇匀,即得。
2.2.3 供试品溶液制备 取复方甘草片10片,称重,研细,精密称取约4片量,置于50 mL量瓶中,精密加提取溶剂(80%甲醇溶液,含0.5%磷酸)50 mL,精密称定,45℃超声处理(功率240 W,频率40 kHz)10 min,静置至室温,再精密称定,用提取溶剂补足减失的重量,摇匀,0.45 μm滤膜滤过,取续滤液,即得。
2.3 色谱条件
色谱柱为COSMOSIL 5C18-MS-Ⅱ柱(250 mm×4.6 mm,5 μm);以0.2%磷 酸 水 溶 液(含0.005 mol·L-1庚烷磺酸钠)为水相A,乙腈-甲醇(9∶1,V/V)溶液为有机相B,梯度洗脱(0~10 min,96%~79%A;10~20 min,79%~65%A;20~32 min,65%~47%A;32~45 min,47%~18%A;45~50 min,18%~15%A;50~55 min,15%~96%A);检测波长为220 nm,柱温为35℃,流速为1.0 mL·min-1,进样量为5 μL。
3 结果与讨论
3.1 方法学考察
在此定量指纹图谱系统的色谱条件下,以甘草酸(CHA)峰为参照物峰,并用压缩因子τ的平方校正理论塔板数后要求应不低于8500[10]。此外,连续进样测定6次考察仪器精密度;精密吸取S1供试品溶液,分别在溶液制备后的0、2、4、6、14、22 h进样测定,考察供试品溶液的稳定性;通过分析6个单独同法制备的S1供试品溶液考察方法的重复性。评价时以CHA峰的保留时间和峰面积为参照,各共有指纹峰的相对保留时间的RSD均小于1.0%,相对峰面积的RSD均小于5.0%,结果表明仪器进样精密度良好,供试品溶液在室温放置22 h内稳定,方法重复性良好。
3.2 复方甘草片指纹图谱建立和评价
按“2.3”项下色谱条件测定9厂家共145批复方甘草片,记录色谱图。将积分后*.cdf文件导入【中药主组分一致性数字化评价系统2.0】软件(带审计追踪功能),以CHA峰的保留时间和峰面积为参照,确定28个共有指纹峰,按平均值法生成标准指纹图谱(RFP),见图2,其中10号峰为MP峰,14号峰为LQN峰,15号峰为MMP峰,16号峰为SB峰,27号峰为CHA峰。以此RFP为评价标准对145批复方甘草片进行评价,样品指纹图谱与RFP的Sm不得低于0.90,Pm应在80%~120%。根据所得评价结果,以Sm和Pm为参数进行聚类分析,采用SPSS软件进行系统聚类,结果S75、S81~S85、S107~S109为第Ⅰ类,其余为第Ⅱ类。虽然Ⅰ类Pm数值偏大,但全部在规定范围内,决定不剔除任何批次样品。9厂家样品定量指纹图谱评价结果见表2(以每个厂家所有批次结果的均值体现),表明9厂家产品质量均合格。其中厂家A生产的批号为20171213、厂家B生产的批号为PDE0712、厂家E生产的批号为3180502、厂家G生产的批号为20180620以及厂家H生产的批号为18053001和18060501的复方甘草片的Sm≥0.95,Pm≈100%,可初步选作复方甘草片的标准制剂[11]。试验中每隔一段时间进样测定双标溶液,记录色谱图,计算相对定量校正因子。结果表明在复方甘草片特征指纹图谱研究期间(约45 d)以及样品测定期间,fqi始终在0.97~1.03,系统定量性质无显著变化,不需对色谱系统进行双标校正。
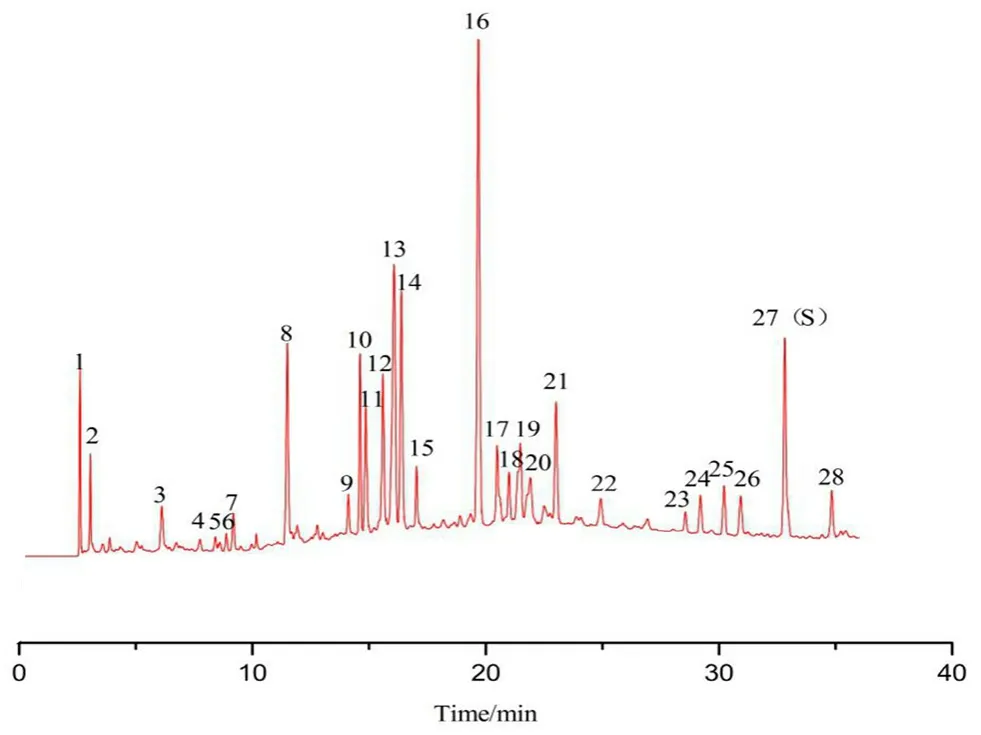
图2 复方甘草片对照指纹图谱(RFP)Fig 2 RFP of compound licorice tablet
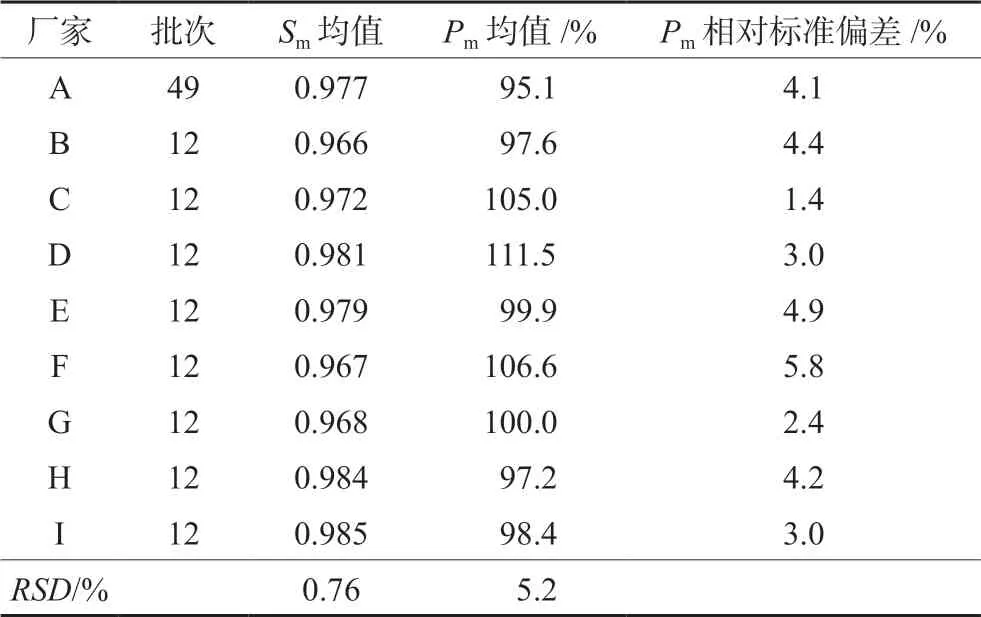
表2 系统指纹定量法评价复方甘草片质量一致性结果 Tab 2 Quality consistency of evaluated by the systematically quantitative fingerprints
从表2的结果可看出,所有厂家样品的平均Sm均大于0.96,说明不同厂家生产的样品之间定性相似度均良好,但只以定性相似度进行评价,不同批次间的差异不显著,不能有效地将不同批次的样品质量区分开来,只能表明不同厂家样品之间的化学指纹数量和分布比例十分相似。因此在评价过程中引入Pm是十分必要的,根据Pm结果可进一步区分不同厂家生产的复方甘草片的质量优劣以及同一厂家样品的批间相似性大小。从结果中可看出厂家A生产的复方甘草片平均Pm值最小,厂家D生产的复方甘草片平均Pm值最大,两者相差16.4%,说明不同厂家样品的化学指纹含量存在显著差异。由此可见,中药质量评价时仅仅采取指纹图谱定性参数评价不能达到全面控制中药质量的目的,定性参数结合指纹图谱的定量相似度参数才能更准确地评价中药质量。
3.3 含量测定
本文采用两种方法测定复方甘草片中5种化合物含量:
① 标准曲线法:精密称定MP、LQN、MMP、SB和CHAA适量,用甲醇稀释,制成6个质量浓度的混合对照品溶液。每份溶液在上述色谱条件下各平行测定两次,以峰面积均值(Aavg)对各对照品浓度(C,mg·mL-1)进行回归,结果见表3。5种化合物在各线性范围内的线性关系均很好。利用各标准曲线以外标法计算145批样品中5种化合物含量,其中CHA含量应在CHAA标准曲线计算所得值基础上乘以0.9797[12]。
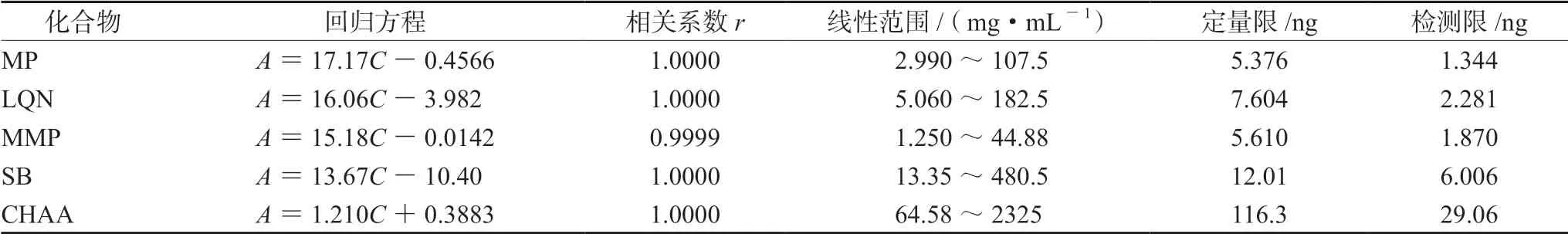
表3 5种化合物线性方程、相关系数、线性范围、定量限和检测限 Tab 3 Linear regression equation,correlation coefficient,linearity,LOQ and LOD of the five Q-markers
② 一线多评法:利用标准曲线根据公式分别计算一测物和多评物的bas/bai与fs/fi,进而计算出各标准曲线的fsi,见表4。以标准曲线法计算SB含量,再以SB含量和fsi计算其他4组分含量。另外,各组分的定量可靠度均不低于96.5%,见表4,表明方法可靠性满足测定要求。以上两种含量计算方法所得结果见表5(以每个厂家各含量结果均值体现)。另外,《中国药典》2020年版要求每片复方甘草片中MP含量在0.36~0.44 mg,CHA含量不少于7.3 mg[12],根据表5结果可知,除厂家A和厂家G的MP平均含量不合格外,其余厂家MP平均含量均符合药典规定,此外,所有厂家CHA平均含量均达到药典标准。
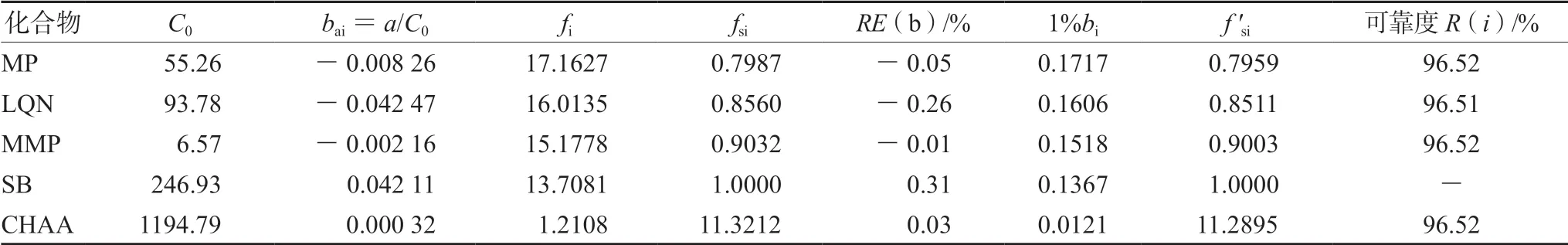
表4 复方甘草片一线多评法的各项参数值 Tab 4 Parameter of compound licorice tablet by multi-markers assay by monolinear method

表5 5种化合物含量测定结果 Tab 5 Content of the five Q-markers
将标准曲线法与一线多评法计算所得的每个厂家每种组分的平均含量结果进行比较,计算相对误差,见表6,两种计算方法所得相对误差最大值为0.46%,表明一线多评法能准确测定复方甘草片中5种化合物含量。相较于标准曲线法,一线多评法可节省对照品与分析时间,为中药及其复方制剂的含量测定提供了一种新的、简便的思路。
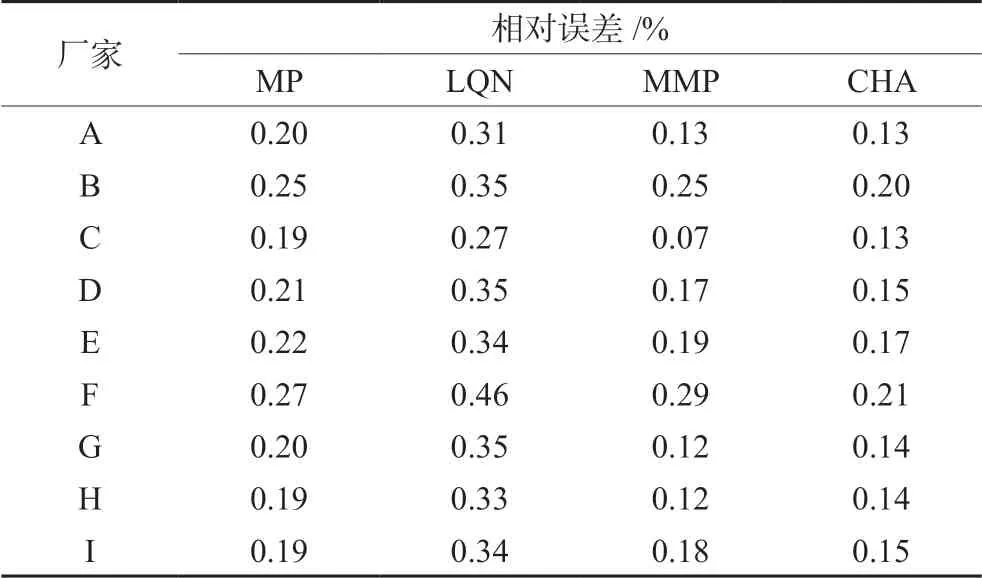
表6 一线多评法与标准曲线法的相对误差 Tab 6 Relative errors between standard curve method and multimarkers assay by monolinear method
4 讨论
本文以复方甘草片为研究对象,通过高效液相色谱法测定145批样品指纹图谱,并建立复方甘草片标准制剂的标准指纹图谱,以系统指纹定量法进行评价。本文提出中药一致性评价应以定量指纹图谱联合一线多评法,实现复方甘草片主组分为中药指纹组分的复方中药制剂的整体质量的量化控制。本文提出一线多评法理论、误差估算方法和可靠度评价方法,该方法主要采用统一化色谱条件,建立双标校正法,消除测定系统变动带来的系统定量误差。通过比较一线多评法和标准曲线法的计算结果,可知一线多评法所得结果准确可靠,操作简便,节省对照品,可降低检测成本和减少分析时间,为中药一致性评价既提供了定量指纹图谱控制方法,也提供了一线多评法的多指标定量方法,达到了在整体上控制好化学指纹物质一致性的目的。因此,中药定量指纹图谱联合一线多评法构成了中药一致性评价的首要关键技术。本课题组在复方甘草片质量一致性评价中充分使用标准制剂控制模型,用价廉易得的苯甲酸钠对照品作为一测物来同时定量复方甘草片中其他4种药效物质,收到理想效果。
猜你喜欢
小哥白尼(趣味科学)(2021年11期)2021-02-28
世界最新医学信息文摘(2020年88期)2020-12-23
小天使·一年级语数英综合(2020年10期)2020-12-16
国学(2020年1期)2020-06-29
华声文萃(2018年11期)2018-07-13
益寿宝典(2018年14期)2018-01-27
数学物理学报(2017年6期)2018-01-22
摄影之友(影像视觉)(2017年1期)2017-07-18
自动化学报(2016年8期)2016-04-16
青少年科技博览(中学版)(2015年7期)2015-08-12
