
帕博西尼关键中间体的合成工艺改进
2022-02-01 07:13石哲吴姗姗霍瑞丽梁迪杨颖
当代化工研究 2022年24期
*石哲 吴姗姗 霍瑞丽 梁迪* 杨颖
(1.吉林大学药学院 吉林 130000 2.吉林省吉测检测技术有限公司 吉林 130000)
帕博西尼(Palbociclib)是美国辉瑞公司研发的一种细胞周期蛋白依赖性激酶(CDK4/6)抑制剂[1]。其通过选择性地抑制CDK4/6,降低CDK4/6的过表达,恢复正常的细胞周期,阻断肿瘤细胞的增殖[2]。临床上,帕博西尼与来曲唑联合用于治疗未曾接受过系统治疗的雌激素受体阳性(ER+)、人表皮生长因子受体2阴性(HER2-)的绝经期女性晚期乳 腺癌[3]。
原研路线制备帕博西尼的关键技术是首先制备吡啶并嘧啶中间体6-溴-8-环戊基-2-甲基亚磺酰基-5-甲基-8H-吡啶并[2,3-D]嘧啶-7-酮(化合物1),在制备得到该中间体的基础上,再经过3步反应制备得到帕博西尼[4]。化合物1的英文名是6-bromo-8-cyclopentyl-2-methylsulfinyl-5-methyl-8H-pyrimidin-7-one,分子式:C14H16BrN3O2S,分子量370.26,化学结构式,见图1。
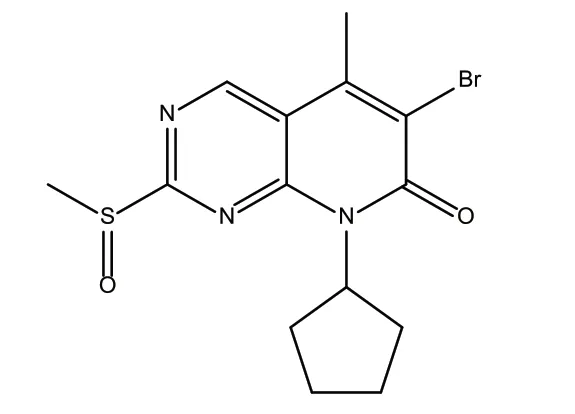
图1 化合物1的化学结构式
原研路线以4-氯-2-甲硫基嘧啶-5-羧酸乙酯为起始原料,与环戊胺在碱性条件下经N-烃化反应得到化合物2,化合物2再经氢化铝锂还原和二氧化锰氧化得到化合物3。化合物3通过格式反应和氧化反应合成化合物4,化合物4经过Witting-horner反应和酰化反应得到化合物5,然后经过溴代和戴维斯氧氮杂环丙烷氧化得到化合物1,具体的工艺路线如图2。该工艺路线存在着反应步骤长,工艺繁琐,主要原料4-氯-2-甲硫基嘧啶-5-羧酸乙酯不易获得,所用试剂如戴维斯氧氮杂环丙烷、氢化铝锂价格昂贵等问题,这些因素限制了该工艺的工业化生产。
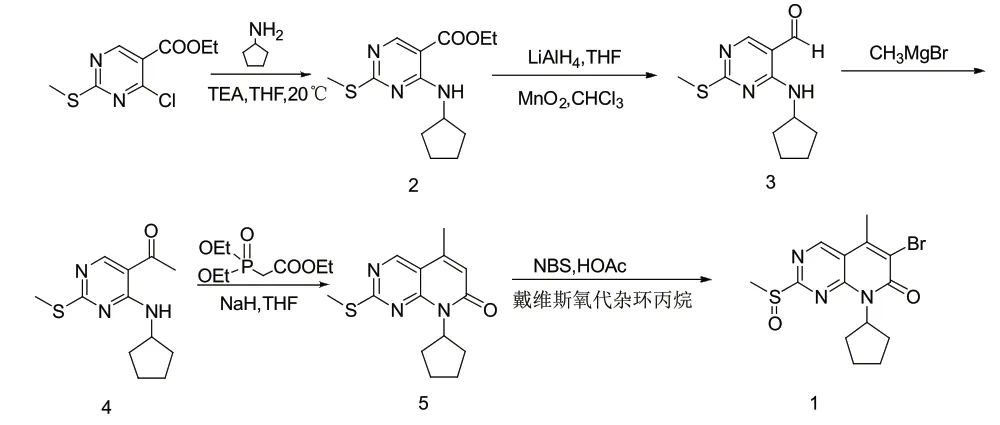
图2 化合物1的原研合成路线
本文对化合物1的制备工艺进行了优化和改进:(1)重新设计了化合物1的合成路线,选择以价格相对便宜的4-氯-2-甲硫基-5-氰基嘧啶为原料,在碱性条件下与环戊胺发生亲核取代反应得到化合物6,化合物6与格式试剂发生甲基化反应得到化合物4,然后经Witting-horner反应得到化合物5,化合物5再在N-溴代琥珀酰亚胺(NBS)作用下发生溴化和氧化得到化合物1,合成路线见图3。该路线降低了工艺成本,缩短了反应步骤,提高了反应收率;(2)对工艺中的主要参数例如溶剂、催化剂、物料摩尔比、反应温度进行了考察优化,提高了反应收率。
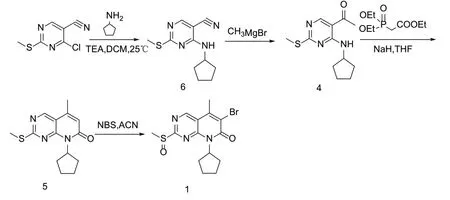
图3 本研究化合物1的合成路线
1.实验部分
(1)仪器和试剂
仪器:AVANCE NEO型核磁共振波谱仪(德国布鲁克公司)、6546 LC/Q-TOF型四极杆飞行时间液质联用系统(美国安捷伦公司)、IRAffinity-1s型红外分光光度计(日本岛津公司)。
试剂:所采用的试剂均属于化学纯或者分析纯,未对其实施纯化处理。
(2)实验步骤
①化合物6的合成
将25.00g(135.0mmol)4-氯-2-甲硫基-5-氰基嘧啶溶于200mL二氯甲烷中,搅拌溶解后于常温缓慢滴加25.20g(148.5mmol)环戊胺和40.90g(405.0mmol)三乙胺溶于50mL二氯甲烷中混匀成的溶液。室温反应3h,TLC监测反应完成后,用100mL水洗3次,100mL饱和氯化钠溶液水洗后分液,合并有机层,无水硫酸钠干燥,过滤、滤液减压浓缩得到白色固体30.00g(收率95%)。ESI-MS m/z:235.1011 [M+H]+;1H-NMR(300MHz,CDCl3)δ:8.21(s,1H),5.64-5.28(m,1H),4.47(q,J=6.9Hz,1H),2.53(s,3H),2.17-2.04(m,2H),1.85-1.72(m,2H),1.70-1.63(m,2H),1.57-1.44(m,2H);13C-NMR(75MHz,CDCl3)δ:159.47 158.76,115.53,52.94,33.03,23.75,14.18。
②化合物4的合成
在氮气保护条件下,将30.00g(128.2mmol)化合物6溶于150mL无水四氢呋喃中,冰水浴冷却至0℃,缓慢滴加甲基溴化镁的四氢呋喃溶液128.2mL(3.0mol/L),滴加完毕,反应2.5h,TLC监测反应。反应完成后,缓慢加入6N盐酸水溶液30mL,升温加热到60℃反应0.5h,减压浓缩除去四氢呋喃后,加入150mL乙酸乙酯并用饱和碳酸氢钠水溶液洗涤至中性,合并有机相,饱和氯化钠水溶液洗涤一次,有机相用无水硫酸钠干燥,减压旋干,得到白色固体27.40g(收率85%)。ESI-MSm/z:252.1164[M+H]+;1H-NMR(300MHz,CDCl3)δ:9.22(s,1H),8.54(s,1H),4.58-4.44(m,1H),2.53(s,3H),2.49(s,3H),2.11-2.02(m,2H),1.83-1.71(m,2H),1.71-1.62 (m,2H),1.62-1.51(m,2H);13C-NMR(75MHz,CDCl3)δ:198.15,175.95,159.23,158.80,108.14,52.07,33.05,26.25,23.79,14.16。
③化合物5的合成
在氮气保护下,将9.95g(248.9mmol)氢化钠分散到300mL无水四氢呋喃中,冷却体系温度至0℃,缓慢滴加55.80g(248.9mmol)磷酰基乙酸三乙酯,约15min滴加完毕。继续搅拌15min后,将25.00g(99.6mmol)化合物4溶于100mL无水四氢呋喃中,缓慢滴加到反应体系,约0.5h滴加完毕,升温至75℃回流20h,TLC监测反应完成后,滴加60mL蒸馏水猝灭反应,冷却至室温,静置1h,抽滤,滤饼用石油醚和水洗涤,真空干燥,得到白色固体25.50g(收率93%)ESI-MSm/z:276.1166[M+H]+;1H-NMR(300MHz,CDCl3)δ:8.69(s,1H),6.43(d,J=1.2Hz,1H),5.90(p,J=8.9Hz,1H),2.63(s,3H),2.41(d,J=1.2Hz,3H),2.38-2.14(m,2H),2.14-1.99(m,2H),1.96-1.79(m,2H),1.77-1.61(m,2H);13CNMR(75MHz,CDCl3)δ:172.11,162.89,153.85,153.82,143.76,121.78,110.73,53.50,28.14,25.56,17.06,14.32。
④化合物1的合成
向反应体系中依次加入25.00g(90.9mmol)化合物5,12.30g(181.7mmol)NBS和300mL乙腈,搅拌均匀后滴加0.55g冰醋酸和1.5mL水,反应2.5h,TLC监测反应完成,将反应液倒入500mL水中,继续搅拌0.5h,抽滤,滤饼用乙醇和水淋洗,在50℃下真空干燥,得到白色固体28.80g(收率86%)。ESI-MSm/z:370.0218[M+H]+;1H-NMR(300MHz,CDCl3)δ:9.09(s,1H),6.23-5.92(m,1H),2.96(s,3H),2.70(s,3H),2.25-2.16(m,2H),2.12(m,2H),1.93(m,2H),1.68(m,2H);13C-NMR(75MHz,CDCl3)δ:172.79,158.06,155.27,154.04,142.58,124.00,114.26,55.69,40.30,28.68,28.58,26.00,25.90,18.43。
2.结果与讨论
(1)化合物1的红外光谱解析
依据红外分光光度法(中国药典2020年版四部通则 0402)对化合物1进行测定,结果如图4。
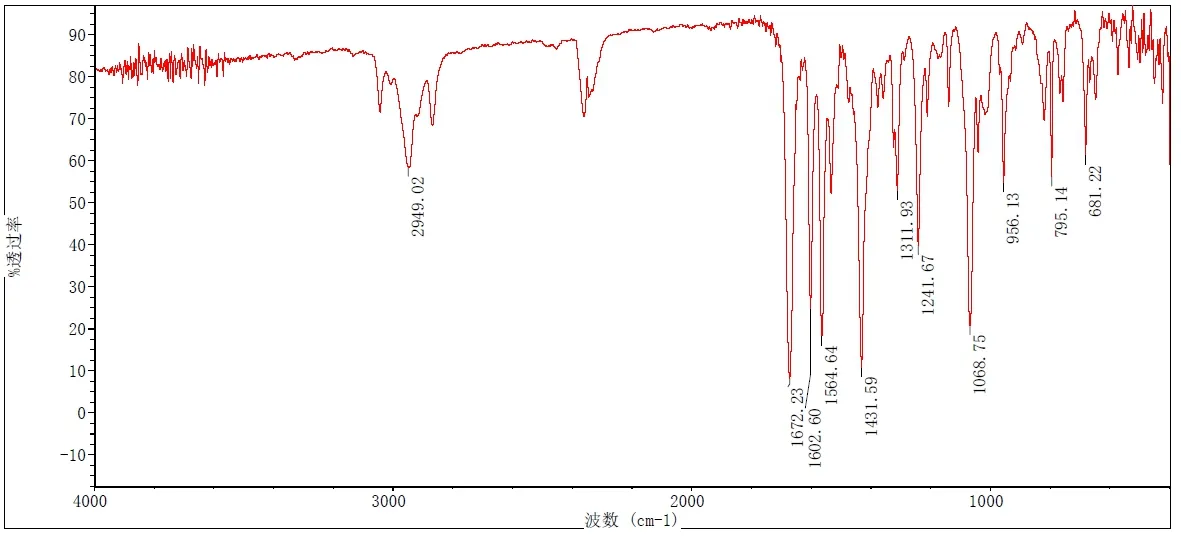
图4 化合物1的红外光谱图
从数据分析,2949.02cm-1的吸收峰,对应为环戊胺基上C-H键的伸缩振动峰;1602.60cm-1的吸收峰,对应为吡啶环上C=O键的伸缩振动峰;1068.75cm-1的吸收峰,对应为亚磺酰基上S=O键的伸缩振动峰。
(2)各步骤的工艺优化
①化合物6的工艺优化
本反应为4-氯-2-甲硫基-5-氰基嘧啶与环戊胺的亲核取代反应,考察了碱、溶剂、环戊胺的投料量和反应温度对产品收率的影响,实验结果,见表1。
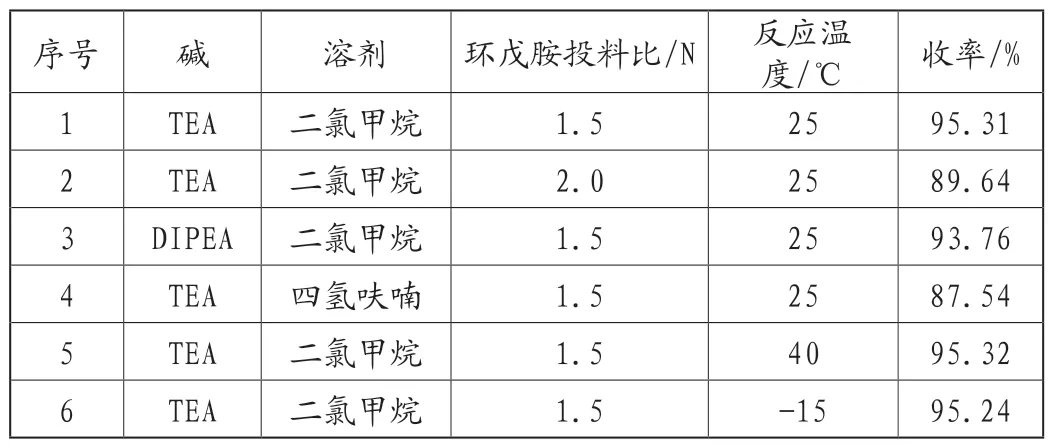
表1 不同反应条件对化合物6收率的影响
由表1可知,有机碱对反应收率的影响不大,考虑成本因素,优选三乙胺;二氯甲烷作为溶剂要优于四氢呋喃;环戊胺投料过多会引起收率降低,推测是由于环戊胺取代了4-氯-2-甲硫基-5-氰基嘧啶2位上的甲硫基,因此优选1.5N;反应温度对反应收率的影响不大,优选25℃条件。
②化合物4的工艺优化
本反应为格式试剂参与的亲核加成反应,控制原料以及溶剂的含水量至关重要,对格式试剂及其滴加速度进行筛选,实验结果,见表2。
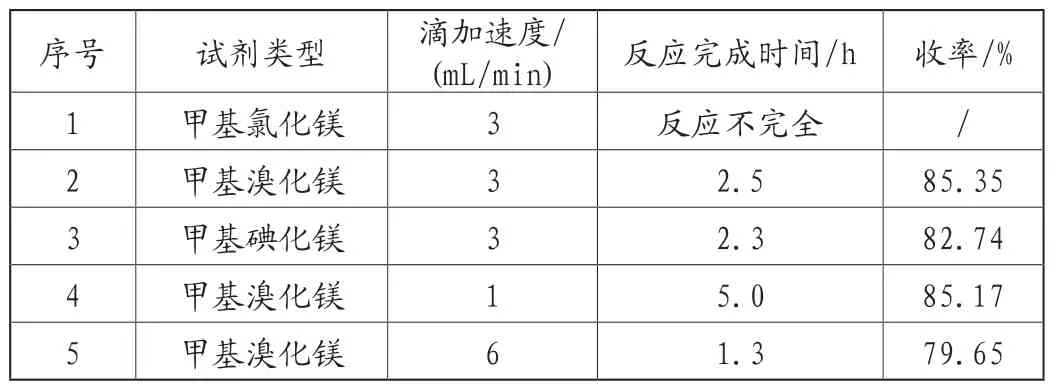
表2 格式试剂类型和滴加速度对产品收率的影响
由表2可知,甲基氯化镁会导致反应不完全,甲基碘化镁虽然会缩短反应时间,但是从成本和收率角度来考虑,优选甲基溴化镁;甲基溴化镁的滴加速度过快会导致收率降低,从时间成本上考虑,优选3mL/min的滴加速度。
③化合物1的工艺优化
该反应为自由基机理的溴代反应,醋酸作为引发剂,常温下即可反应完全,由于NBS较为活泼,可以通过控制NBS的量来避免产生较多的二溴代杂质,实验结果,见表3。
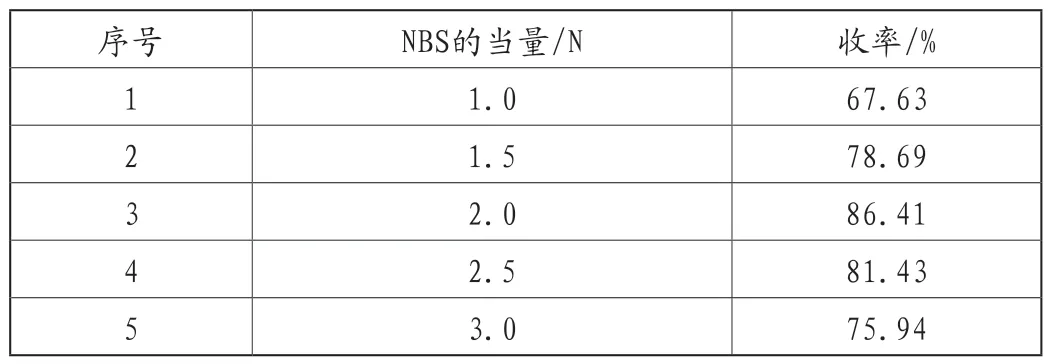
表3 NBS的量对产品收率的影响
从表3可以看出,当NBS的当量为2.0N时,反应的收率较高,同时在反应过程中发现,当NBS的当量为1.0N时,无法反应完全,但是当NBS的当量为3.0N时,收率反而降低,推测可能是产生了二取代杂质,因此优选NBS的投料量为2.0N。
3.结论
综上所述,本研究对帕博西尼重要中间体6-溴-8-环戊基-2-甲基亚磺酰基-5-甲基-8H-吡啶并[2,3-D]嘧啶-7-酮的原研合成路线进行了优化,采用4-氯-2-甲硫基-5-氰基嘧啶为主要原料,经过亲核取代反应、Witting-horner反应、溴代和氧化反应制备了目标产物;并对合成过程中的关键参数进行了优化,产品收率为64.58%,优于现有技术工艺。该工艺使得原本复杂的流程得到了简化,反应条件更为温和,更加适合推广运用。
猜你喜欢
世界农药(2022年10期)2022-11-10
当代化工研究(2022年11期)2022-06-27
中国酿造(2022年1期)2022-02-07
能源化工(2021年2期)2021-12-30
昆明医科大学学报(2021年8期)2021-08-13
昆明医科大学学报(2021年3期)2021-07-22
农药科学与管理(2019年6期)2019-11-23
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
应用化工(2015年12期)2015-04-14
浙江化工(2014年9期)2014-08-15
