
UPLC-MS/MS法测定美洛昔康中两个中间体杂质
2022-02-24 11:55葛雪松张新勇
广州化工 2022年3期
孙 桐,葛雪松,张新勇,卓 静
(宿迁市食品药品检验所, 江苏 宿迁 223800)
美洛昔康(Meloxicam),化学名4-羟基-2-甲基-N-(5-甲基-2-噻唑基)-2H-1,2-苯并噻嗪-3-羧酰胺1,1-二氧化物,是一种非类固醇解热镇痛抗炎药。由德国Boehringer Ingelheim公司于1996年首次在南非上市,具有镇痛、抗炎、解热作用,临床适用于类风湿性关节炎和疼痛性骨关节炎的对症治疗。根据美洛昔康原料药的生产工艺[1]分析,4-羟基-2-甲基-2H-1,2-苯并噻嗪-3-羧酸乙酯-1,1-二氧化物(杂质A)和2-氨基-5-甲基噻唑(杂质B)是美洛昔康合成工艺中两种重要的中间体,极有可能残留在原料药中,最终传递到成品中[2-5]。杂质A,B均有一定的毒性,而在现行的质量控制标准中,仅仅使用供试品稀释的自身对照法对杂质进行控制,具有很大的局限性。参照美国药典USP43-NF38[6]和欧洲药典EP10.0[7],原料药中均对杂质A,B进行了单独的限度控制。为了大众用药安全,需严格控制原料药质量,结合相关文献[8-16],建立了LC-MS/MS法同时测定美洛昔康中杂质A和杂质B。杂质A,B的结构式见图1。
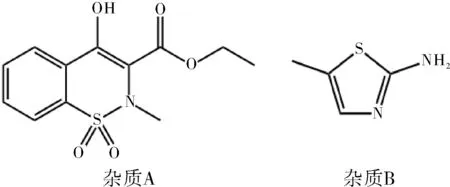
图1 杂质A,杂质B结构图
1 仪器与试药
1.1 仪 器
XEVO TQ-S micro三重四级杆液质联用色谱仪,美国Waters;XP205电子天平(0.01 mg),瑞士梅特勒,Milli-Q超纯水机,美国Millipore。
1.2 试 药
美洛昔康对照品(批号:100679-201903,含量100%),中国食品药品检定研究院;美洛昔康杂质A对照品(批号:0301-RC-0087,含量100%),美国CATO;美洛昔康杂质B对照品(批号:0223-RC-0093,含量95.7%),美国CATO;美洛昔康原料药(批号:20210318,20210319,20210320),国内某药业有限公司,甲醇(色谱纯),德国Merk、甲酸(色谱纯),美国ACS,水为自制超纯水。
2 方法与结果
2.1 色谱与质谱条件
2.1.1 色谱条件
采用ACQUITY UPLC©HSS T3 C18(2.1 mm×100 mm, 1.8 μm)色谱柱,流动相为0.1%甲酸水溶液-甲醇(45:55),流速为0.3 mL/min,柱温35 ℃,进样盘温度10 ℃,进样体积1 μL。
2.1.2 质谱条件
采用电喷雾离子源(ESI),多反应离子监测模式(MRM),正离子扫描,脱溶剂温度:500 ℃,干燥气流速:15 L/min,毛细管电压:2.5 kV,质谱监测时间:0~5.9 min(系统适用性条件下监测时间:0~8 min),其他质谱条件见表1,表1中加*的为定量离子。
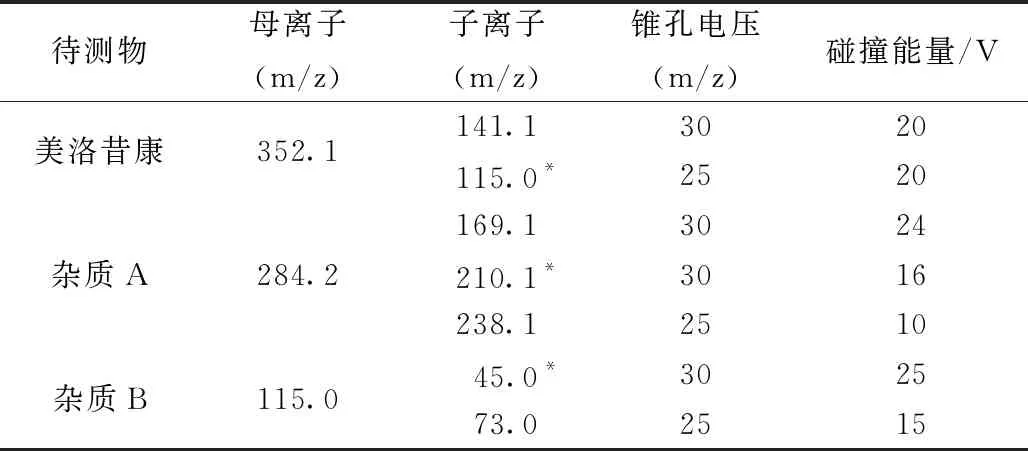
表1 质谱分析参数
2.2 溶液的配制
2.2.1 供试品溶液
精密称取美洛昔康原料药约50 mg,置50 mL量瓶中,加甲醇溶解并稀释至刻度,摇匀,即得美洛昔康的供试品溶液。
2.2.2 混合对照品溶液
分别精密称取美洛昔康,杂质A和杂质B的对照品适量,用甲醇溶解并稀释至每1 mL分别含美洛昔康,杂质A和杂质B约1 μg的溶液,作为单一对照品贮备液①,②,③;精密量取单一对照品贮备液①,②,③适量,用70%甲醇水稀释至每1 mL含美洛昔康,杂质A和杂质B 均约为0.1 μg的系统适应性溶液;精密量取单一对照品贮备液②,③适量,用70%甲醇稀释至每1 mL含杂质A和杂质B均约为0.1 μg的混合对照品贮备液。
2.3 方法考察
2.3.1 专属性实验
取单一对照品贮备液①,②,③各 0.1 μL,空白溶剂(70%甲醇水),系统适应性溶液,混合对照品贮备液和供试品溶液各 1 μL,分别注入液质联用仪,记录质谱图和色谱图,见图2,3。结果显示,各成分保留时间:杂质B 0.72 min,杂质A 5.13 min,美洛昔康6.18 min;空白溶剂对杂质无干扰,各色谱峰之间无干扰。

图2 美洛昔康及各杂质的质谱图
2.3.2 线性关系考察
精密量取“2.2.2”项下的混合对照品贮备液,用70%甲醇水稀释成含杂质A 1.03,2.06,4.12,10.32,20.64,51.50,100.30 ng/mL;杂质B 0.98,1.96,3.92,9.80,19.60,49.0,98.0 ng/mL的混合对照品溶液。取上述混合对照品溶液,分别进样,记录色谱图,以峰面积Y为纵坐标,溶液浓度X(ng/mL),进行线性回归。杂质A,B的线性方程如下:
这一年图科回归,总经理叶鹏又选来了一个瘦高灵活,能里能外的外援拉米扎纳。这两人特点互补性格开朗,每场都能给对手带来强大的压力,人称“黄金双枪”。
Y=393.858X-200.006(r=0.9992),杂质A在1.03~100.3 ng/mL的范围内线性关系良好
Y=1087.44X-402.125(r=0.9996),杂质B在0.98~ 98.0 ng/mL的范围内线性关系良好。
2.3.3 定量限和检出限
取“2.2.2”项下的混合对照品贮备液,用70%甲醇水,逐级稀释,稀释到杂质A,B峰的信噪比(S/N)约为10的时候,溶液浓度作为定量限;信噪比(S/N)约为3的时候,溶液浓度作为检出限。实验结果:杂质A的检出限0.31 ng/mL,定量限0.83 ng/mL;杂质B的检出限0.15 ng/mL,定量限0.51 ng/mL。
2.3.4 精密度试验
取线性考察下混合对照品溶液(杂质A 10.32 ng/mL,杂质B 9.80 ng/mL),连续进样6次,以峰面积计算各杂质的RSD,杂质A的RSD=2.6%(n=6);杂质B的RSD=1.8%(n=6),结果表明方法的精密度良好。

图3 空白溶剂(A), 系统适应性溶液(B), 混合对照品(C)及供试品(D)的色谱图
2.3.5 中间精密度试验
由另一位实验人员在同一台液质联用仪上做精密度试验,结合两者的结果计算RSD,杂质A的RSD=3.7%(n=12);杂质B的RSD=2.9%(n=12)。
2.3.6 重复性试验
2.3.7 稳定性试验
配置混合对照品溶液(杂质A,B浓度均约为100 ng/mL),在室温(20 ℃)情况下放置0,2,4,6,8后进样分析,以峰面积计算。结果显示:杂质A的RSD=3.1%(n=5),杂质B的RSD=1.9%(n=5),说明杂质A,B在8 h内稳定性良好。
2.3.8 耐用性试验
取美洛昔康原料药按“2.2.1”项下的方法平行配置2份供试品溶液,分别在色谱柱温度(33 ℃和37 ℃),流动相比例[0.1%甲酸水溶液:甲醇(43:57),0.1%甲酸水溶液:甲醇(47:53)],流动相流速(0.29,0.31 mL/min)下进样分析,计算各杂质含量,与原有色谱条件下的含量进行比较。结果显示,当色谱条件在温度,流速,流动相比例细微变化的情况下,各杂质含量的RSD<10%,说明该方法的耐用性良好。
2.3.9 加样回收率
精密称取杂质A,B对照品适量,用70%甲醇水稀释至每1 mL含杂质A 0.207 μL,杂质B 1.016 μL的混合对照品溶液;精密称取美洛昔康原料药(批号:20210318)约50 mg,共9份,分成三组,分别置50 mL量瓶中,再取上述混合对照品4 mL (3份),5 mL(3份),6 mL(3份)置已加入美洛昔康的量瓶中,用70%甲醇水溶解并稀释至刻度,摇匀,进样分析,计算加样回收率,结果见表2。
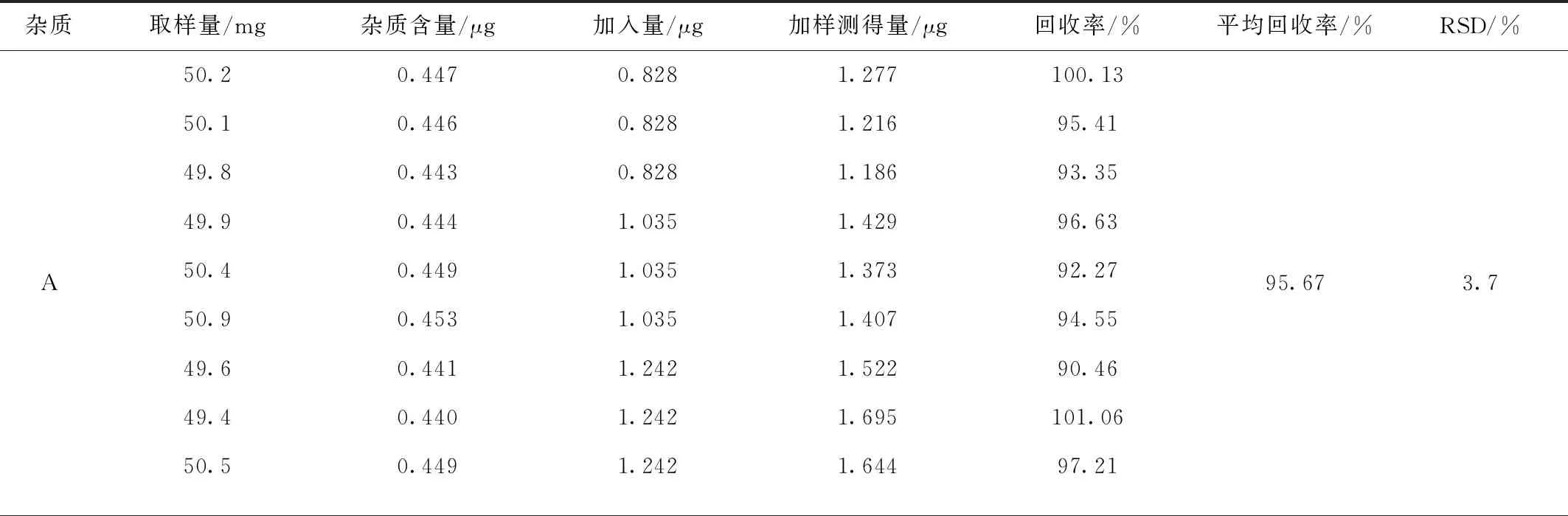
表2 杂质A,B加样回收率结果(n=9)
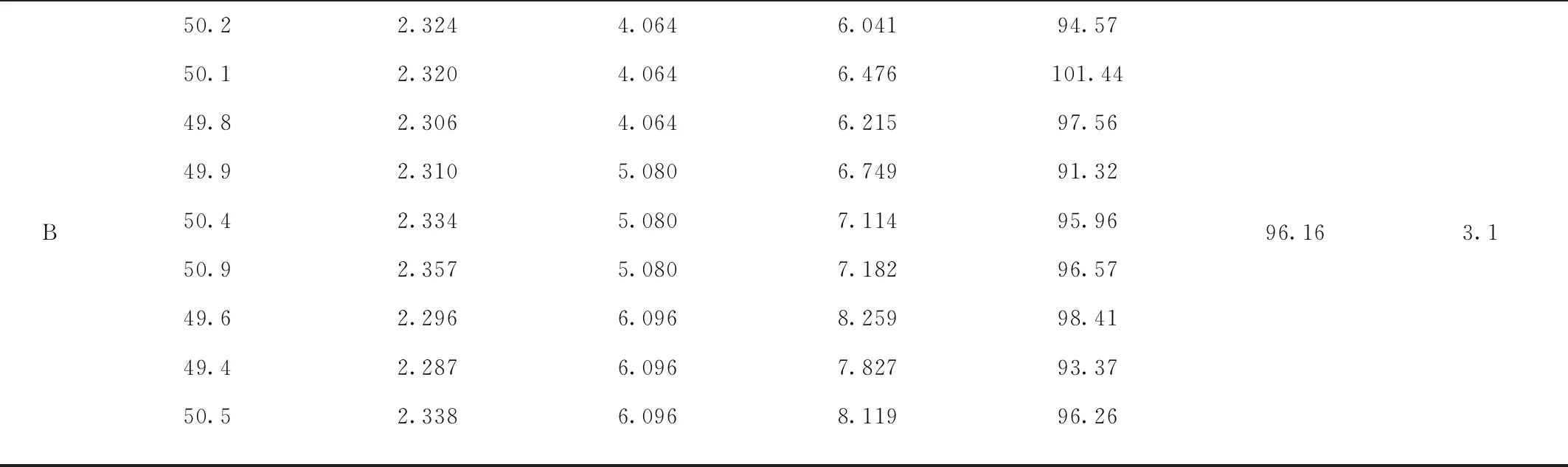
续表2
2.4 样品含量测定
取美洛昔康原料药(20210318,20210319,20210320),按照“2.2.1”项下的方法配置供试品溶液,按“2.1”项下的液质联用条件进样,计算杂质含量,结果见表3。
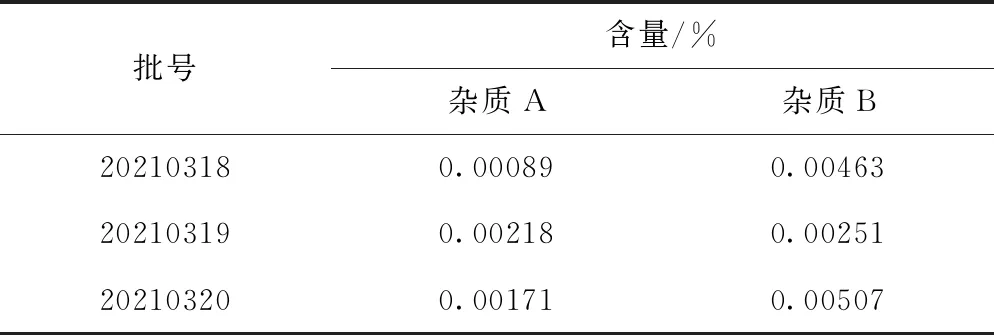
表3 杂质含量测定结果
3 讨 论
3.1 系统适应性要求
供试品溶液主成分美洛昔康含量过高,建议让美洛昔康出峰在最后,通过控制质谱监测时间在美洛昔康出峰时间之前,仅记录杂质A,B的色谱图,让美洛昔康直接以废液形式排出,以免污染质谱检测器。
3.2 流动相的选择
杂质A和B在ES+模式下离子响应强度是ES-模式下的数倍,流动相中加入甲酸铵或乙酸铵抑制杂质A和B的电离;相反,流动相中引入甲酸,有助于提供H离子,增强待测离子的强度。所以本文着重考察了甲醇-甲酸水,乙腈-甲酸水两种常见的流动相体系。乙腈-甲醇水系统,基线噪音偏高,杂质B有明显拖尾,美洛昔康峰的保留时间在杂质A和B之间,不适合。杂质B对酸灵敏,在pH值2~4情况下,离子响应强度明显提高,0.1%的甲酸完全满足实验要求。调整甲醇-甲酸水体系流动相比例,高比例水相情况下,杂质B峰型差,峰展宽偏大,有明显拖尾;高比例有机相时,杂质A与美洛昔康的分离效果变差,通过反复调整,最终确定流动相比例为甲醇:0.1%甲酸水=55:45。
3.3 杂质B同为降解产物的验证
由图2可见,美洛昔康的子离子(m/z=115.0)与杂质B的母离子(m/z=115.0)质量数一致,推测杂质B也是美洛昔康的降解产物。美洛昔康对酸碱稳定,因此对美洛昔康原料药分别进行高温,光照,过氧化氢溶液氧化的破坏性试验,然后进样分析杂质B的含量,结果显示,在高温,光照下杂质B含量有少量提高,氧化破坏下杂质B含量提高了几十倍,破坏性试验验证了杂质B也是美洛昔康的降解产物。
3.4 杂质A和B的限度
杂质A和B未有完整的毒性毒理研究报告,通过资料文献仅了解二者都是吸入有毒,杂质B对皮肤,粘膜有刺激作用,对眼睛刺激强烈,但可以排除基因杂质的可能性。参考化学药物杂质研究技术指导原则和ICH相关规定,同时结合美国药典USP43-NF38和欧洲药典EP10.0中美洛昔康原料药有关物质的质量规定 ,建议将杂质A和B的限度均定为0.1%。
4 结 语
随着科技的进步,药品质量也在逐步提高。药品质量的提高关键在有关物质,对药物有关物质中各类杂质的控制,显著的影响着药物的安全和质量。现如今,中国药典中多数药物还是采用主成分稀释自身对照法,对单个和杂质总量进行控制,这么笼统的控制是不够的,每个杂质的毒性,危害性不尽相同,我们需要对不同的杂质进行系统分析,进而做出单独的限度控制。在杂质分析领域,LC-MS/MS有着专属性强,灵敏度高,精密度高,检测迅速等诸多优点,还避免了样品复杂的前处理过程,已成为药品中微量杂质分析的首选技术。方法学验证结果证明,该方法可以有效的对杂质A和B进行定性定量分析,为美洛昔康有关物质的质量提高提供参考。
猜你喜欢
核化学与放射化学(2022年2期)2022-04-28
中国经济周刊(2021年22期)2021-12-07
艺术品鉴(2020年6期)2020-12-06
环境保护与循环经济(2020年12期)2020-03-01
环球时报(2020-02-20)2020-02-20
传媒评论(2019年6期)2019-10-14
中国经济信息(2017年17期)2017-09-09
领导文萃(2017年6期)2017-03-24
中学生数理化·高一版(2016年7期)2016-12-07
中学生数理化·中考版(2015年12期)2015-09-10
