
PI3K/AKT信号通路与艾滋病相关的研究进展①
2022-03-22 12:53郑文锦李志慧河南中医药大学郑州450046
中国免疫学杂志 2022年4期
郑文锦 冯 龙 李志慧(河南中医药大学,郑州 450046)
3-磷酸肌醇激酶(phosphoinositide3-kinase,PI3K)/丝、苏氨酸蛋白激酶(protein kinase B,PKB 或AKT)通路在广泛的人类肿瘤谱中失调,在恶性肿瘤的发生和发展中发挥重要作用,参与抑制细胞凋亡、加快细胞周期运行、促进血管形成和肿瘤侵袭转移等过程[1]。不同类型的肿瘤在该通路上均有大量研究,如:肝细胞癌、胃癌、胰腺癌、结肠癌等[2]。PI3K/AKT 信号通路被许多类型的细胞刺激或毒性损伤激活,并调节基本的细胞功能,如转录、翻译、增殖、生长和存活[3]。PI3K/AKT信号通路图如图1。
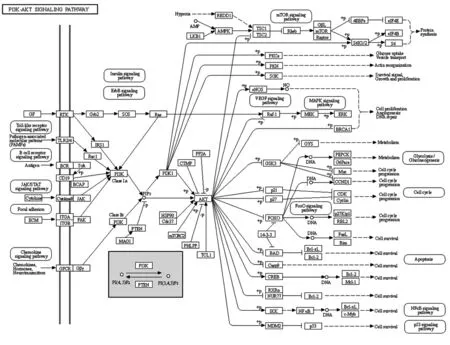
图1 PI3K/AKT信号通路图(引自:KEGG生物信息数据库,https://www.genome.jp/pathway/map04151)Fig.1 PI3K/AKT signaling pathway(Quoted:Kyoto Encyclopedia of Genes and Genomes,https://www.genome.jp/path⁃way/map04151)
PI3K/AKT 通路不仅在肿瘤相关疾病中发挥重要作用,在获得性免疫缺陷综合征(acquired immuno⁃deficiency syndrome,AIDS),即艾滋病的相关研究中也极其重要。AIDS是一种危害性极大的传染病,由人类免疫缺陷病毒(human immunodeficiency vi⁃rus,HIV)感染引起。自1983 年分离出HIV-1 以来,AIDS 在全世界范围内迅速蔓延,威胁人类生命健康[4]。国家卫生健康委员会疾病预防控制局提供的数据显示,我国AIDS 发病数及病死数逐年攀升,2019 年1 月至7 月中国AIDS 发病数为39 483 例,病死人数为11 454 例;2018 年中国AIDS 发病数为64 170例,病死人数为18 780例。
1 PI3K
PI3K是肌醇与磷脂酰肌醇(phosphatidyl inositol,PI)的重要激酶,由其活化产生的类脂产物3,4,5-三磷酸磷脂酰肌醇[PI(3,4,5)P3]作为细胞内的第二信使,是AKT 转位于胞膜并被活化所必需的。PI3K与AKT 组成的PI3K-AKT 信号通路在细胞的增殖和存活中发挥重要作用[5]。
2 AKT
AKT 是丝/苏氨酸蛋白激酶,有AKT1、AKT2、AKT3 3 种亚型,均参与下游底物的活化,其中以AKT2的作用为主。PI3K激活的AKT可通过磷酸化作用激活或抑制其下游靶蛋白Bad、Caspase9、NF-κB、Forkhead、mTOR、Par-4、P21 等,从而介导胰岛素、多种生长因子等诱发的细胞生长,经多种途径促进细胞存活,是重要的抗凋亡调节因子[6]。
3 PI3K/AKT信号通路与HIV-1相关的研究
3.1 单核-巨噬细胞
3.1.1 巨噬细胞的自噬作用 自噬是细胞通过自噬体形成并可与溶酶体融合降解细胞器等胞内物质的精细过程,宿主以此途径防御病原微生物的入侵,包括选择性地将病原运送至溶酶体及将微生物核酸和抗原运送到溶酶体部位以激活宿主免疫反应[7]。病毒感染过程中,巨噬细胞既可对病毒实体直接吞噬清除,也可通过吞噬病毒感染后诱导凋亡的宿主细胞来减轻病毒感染的病理损伤[8]。
针对巨噬细胞对HIV-1 的自噬作用,有研究表明:双重磷脂酰肌醇3-激酶/雷帕霉素(PI3K/mTOR)抑制剂达科利西布(NVP-BEZ235)、PI3K/mTOR/含溴结构域的蛋白4(BRD4)抑制剂SF2523 等都通过自噬降解细胞内的HIV,以剂量依赖的方式降低了巨噬细胞中HIV 的复制。且PI3K/mTOR 和PI3K/mTOR/BRD4 抑制剂抑制HIV 的机制需要自噬体的形成,以及它们随后成熟为自溶体[9]。
3.1.2 巨噬细胞的凋亡 HIV-1 感染后的巨噬细胞部分发生凋亡。AKT-1是巨噬细胞存活的关键蛋白,AKT-1 的过表达增加了转录因子FOXO3a(AKT下游通路蛋白)的磷酸化。具有组成型活性的FOXO3a的过表达增加了MDM 中DNA 的片段化,同时降低了细胞活力[10]。AKT 的活化会导致巨噬细胞凋亡增加,免疫细胞凋亡的调控成为针对免疫学检查点治疗的新角度[11]。
3.1.3 巨噬细胞寿命的延长 感染了HIV-1 的巨噬细胞表现出寿命延长,这与其作为HIV-1 长寿命储存库的作用一致。HIV-1 的这种细胞保护作用有助于人类巨噬细胞贮藏库的长期存活和持久性HIV-1 产生。HIV-1 通过PI3K 信号通路促进细胞存活并防止细胞凋亡[12]。在对其机制的研究中,HILLEBRAND 等[13]认为PI3K 信号通路影响了丝氨酸-精氨酸(SR)蛋白的磷酸化(SR 属于平衡剪接位点选择的剪接调节因子家族)。HIV-1 也可能通过调节剪接调节蛋白的磷酸化来促进PI3K 转录物的加工,进而通过影响PI3K信号通路达到促进宿主细胞存活的目的。
PI3K/AKT 通路不仅对巨噬细胞的寿命延长有作用,以PI3K/AKT 为靶点的药物能够抑制HIV-1病毒的产生。有研究发现HIV-1感染激活了原代人巨噬细胞中的PI3K/AKT 途径,如减少的PTEN 蛋白(AKT 负调控因子)表达和增加的AKT 激酶活性,其结果证明PI3K/AKT 抑制剂(包括临床上可用的Miltefosine)可显著减少长寿病毒感染的巨噬细胞产生的HIV-1[14]。5,7-二羟基-6-甲氧基黄酮可能通过抑制PI3K/AKT 信号通路来消除HIV-1 感染的细胞保护巨噬细胞,并通过缩短感染巨噬细胞寿命在体内发挥抗HIV-1的作用,表明PI3K/AKT 磷酸化抑制剂可以作为有效的抗病毒药物[15]。AKT 抑制剂(perifosine和edelfosine)特异性诱导了被HIV-1感染的原代人类巨噬细胞凋亡。此外,研究还表明periposine 可有效降低HIV-1 感染的原代人类巨噬细胞的病毒产生[16]。
中药提取物在抑制巨噬细胞寿命延长上也有积极作用。人参皂苷Rb1 可通过抑制PI3K/AKT 信号通路消除感染HIV-1 的细胞保护性巨噬细胞[17]。远志科也通过PI3K/AKT 信号通路有效地消除了HIV-1 Tat 转导的CHME5 巨噬细胞的细胞保护作用[18]。
3.2 星形胶质细胞、小胶质细胞与神经认知障碍HAND 在感染了HIV-1 的患者中,与HIV 相关的HAND 患病率仍然很高。被暴露于病毒蛋白的星形胶质细胞/小胶质细胞产生促炎细胞因子被认为是导致HIV-1介导的神经毒性的机制之一。通过使用不同的病毒蛋白诱导神经炎症因子产生,使用不同的药物均得出PI3K/AKT信号通路参与了HAND,见表1。PI3K/AKT 通路除了参与各种神经炎症因子的产生和介导炎症反应,还参与了其他与HAND 相关靶标的研究。HIV-1 Tat 引起星形胶质细胞质膜中兴奋性氨基酸转运蛋白2(EAAT-2)的减少导致细胞外谷氨酸水平升高,进而导致神经元凋亡,PI3K抑制剂或可通过该机制表现出治疗HAND 的作用[23]。另外有研究表明,Tat会导致血小板衍生生长因子-CC(PDGF-CC)的内在表达降低,而用PDGF-CC预处理SH-SY5Y 细胞可通过减轻细胞凋亡和神经突及MAP-2 的丧失来消除Tat 介导的神经毒性,PI3K/AKT 信号传导在PDGF-CC 介导中起神经保护中作用[24]。研究认为HIV-1 Tat增强嘌呤能P2Y4受体信号传导,通过PI3K/AKT 和ERK/MAPK 信号通路(MAPK 信号通路位于PI3K/AKT 通路下游)介导炎症细胞因子产生和神经元损伤[25]。

表1 PI3K/AKT参与HAND的研究进展Tab.1 Research progress of PI3K/AKT participating in HAND
3.3 CD4+T细胞
3.3.1 HIV-1 进入CD4+T 细胞 以防止HIV-1 入侵靶细胞为靶点进行的抗HIV-1研究一直都是热点方向。进入抑制剂包括HIV 吸附抑制剂、HIV 受体CCR5/CXCR4 拮抗剂、融合抑制剂等[26]。研究发现PI3K p110α 亚型特异性抑制剂PIK-75 抑制HIV-1进入HIV-1 允许的T 细胞、PM1 细胞和TZM-bl 细胞(HeLa 细胞来源的指示细胞,共表达CD4、CCR5 和CXCR4),并降低了HIV-1 诱导的AKT 磷酸化。此外,PTEN(AKT 的负调节因子)是HIV-1 进入的关键调节因子。在PI3K p110α 特异性抑制剂的存在下,HIV-1 env-CD4 相互作用引起的细胞间融合受到抑制。这些数据表明,PI3K p110α/PTEN 信号通路对于HIV-1 进入包括HIV-1 env 介导的细胞间融合是至关重要的[27]。在此研究之前,就有针对PI3K抑制剂对HIV 感染的抑制作用的研究,结果显示,PI3K抑制剂阻断了HIV 感染、核导入和T 细胞激活,对TLR2/IL-2 激活的CD4+T 细胞的细胞周期进程有中度抑制作用[28]。
3.3.2 HIV-1 整合和建立潜伏病毒库 研究显示,在静止的CD4+T 细胞中,趋化因子CCL19 诱导了AKT、NF-κB、细胞外信号调节激酶(ERK)和p38 的磷酸化。抑制PI3K和Ras/Raf/促分裂原活化的蛋白激酶/ERK 激酶(MEK)/ERK 信号通路可抑制HIV 整合,但不会显著减少HIV 核进入。HIV 在CCL19 处理的静息CD4+T 细胞中的整合取决于NF-κB 信号传导,且增加了HIV 整合酶的稳定性,从而可以进行后续整合和建立潜伏期。因此SALEH 等[29]认为HIV 整合和CCL19 处理的静息CD4+T 细胞潜伏期的建立需要激活NF-κB。
KIM 等[30]认为感染HIV-1的CD4+T细胞通过活化AKT[HIV-1 Tat蛋白可干扰PTEN(AKT 的负调节因子)为活化AKT 的机制之一],然后通过Bcl-2 家族成员Bad的直接抑制性磷酸化或通过转录因子如FOXO1 的磷酸化间接干扰促凋亡分子(Bad 及FOXO1 均在PI3K/AKT 信号通路下游),所述转录因子随后转移出细胞核,从而阻止促凋亡基因的转录。
3.3.3 病毒储存库的逆转剂 联合抗逆转录病毒疗法(cART)可有效抑制HIV-1 复制,但静止记忆CD4+T 细胞中的潜在病毒库不能被cART 清除,病毒储存库成为治愈HIV-1感染的主要障碍。研究发现PI3K 激动剂57704(对p110α 同工型具有特异性,但对p110β、δ 或γ 同工型没有特异性)通过PI3K/AKT 信号通路重新激活潜在的HIV-1,强调了PI3K/AKT途径参与维持HIV-1潜伏期[31]。
最近研究发现,外来体[一种未感染细胞的细胞外囊泡(EV)]激活了潜伏感染细胞中HIV-1 的转录,而与cART 无关。在这项研究中探讨了潜在HIV-1 的EV 激活HIV-1 的具体机制。研究发现磷酸化的c-Src 存在于各种细胞系的EV 中,并具有激活下游蛋白(例如EGFR)的能力,从而启动信号级联。然后EGFR 能够激活PI3K/AKT/mTOR 途径,从而激活STAT3和SRC-1,最终逆转HIV-1潜伏期[32]。
在治疗浓度下,组蛋白去乙酰化酶(HDAC)和PI3K 的双重抑制剂——抑瘤素(CUDC-907)是一种有效的HIV-1 潜伏期逆转剂,且其不诱导T 细胞活化和增殖[33]。六亚甲基双乙酰胺(HMBA)是慢性感染细胞中细胞分化和HIV产生的有效诱导剂。HMBA瞬时激活PI3K/AKT 途径,从而导致HEXIM1 磷酸化,释放出活性正转录延伸因子b(P-TEFb)。随后,P-TEFb 被募集到HIV 启动子以刺激转录延伸和病毒产生[34]。MAPK p38α(位于PI3K 信号通路下游)可通过激活TCR 相关通路,上调包括PI3K/AKT 在内的多种信号通路蛋白的表达,抵抗HIV-1 潜伏感染中AZT 抑制的HIV-1 复制,并可用作潜伏逆转剂[35]。
研究表明,受感染的CD4+T 细胞与树突状细胞(dendritic cell,DC)的相互作用进一步激活了潜伏的HIV-1。VAN MONTFORT 等[36]认为与单纯TCR刺激相比,接受TCR+DC 刺激的AIDS患者的CD4+T细胞更频繁地逆转潜伏期。这种“组合拳”策略似乎是清除病毒储存库的理想策略,并且研究确定了DC 接触激活CD4+T 细胞中的PI3K/AKT/mTOR通路。
3.4 DC
3.4.1 DC与IFN-α消除 DHAMANAGE 等[37]在最近感染的HIV 患者中观察到IFN-α 消除现象,研究发现暴露于HIV-1 的浆细胞样树突状细胞(plasma⁃cytoid dendritic cell,pDC)没有显示IRF-7 易位到核中。此外,还观察到HIV-1抑制pDC 中PI3K/AKT 信号通路的AKT 磷酸化,这是IRF-7 易位至细胞核的重要步骤。即使在存在Toll 样受体7 激动剂刺激的情况下,HIV-1 诱导的AKT 磷酸化和IRF-7 易位抑制也很明显。这些发现表明,HIV-1 可能抑制IRF-7向pDC 核的转运,从而导致IFN-α 抑制,这些均与PI3K/AKT信号通路有关。
3.4.2 DC与免疫逃逸机制 生殖器和直肠黏膜中未成熟的树突状细胞(iDC)可能是在病毒性传播期间最早与HIV-1 接触的细胞之一。游离的HIV-1 导致感染增加和病毒的免疫逃逸机制可能有PI3K 通路的激活和病毒激活宿主补体系统,导致灭活的补体片段(如iC3b)参与病毒调理。实验研究了暴露于游离HIV-1(F-HIV)、补体调理的HIV-1(C-HIV)及补体和非调理化HIV-1(CI-HIV)后在人iDC 中诱导的抗病毒和炎症反应,F-HIV 诱导的应答是TLR8依赖性的,随后激活了IFN 调节因子1、p38、ERK、PI3K 和NF-κB 途径,TLR8 信号转导的这种调节是由补体受体3 介导的,并导致感染增加。补体系统的病毒劫持对iDC 功能的影响可能是HIV-1 在宿主中建立感染的重要免疫逃逸机制[38]。
4 总结及展望
PI3K/AKT 信号通路不仅在HIV-1 主要的靶细胞(单核-巨噬细胞细胞、胶质细胞、CD4+T 细胞、DC)中发挥作用,还对受HIV 影响的骨髓间充质干细胞、肾小管细胞(HIV 相关肾病,HIVAN)、心肌细胞产生影响[39-41]。PI3K/AKT 信号通路通过在HIV-1几种靶细胞中发挥重要作用而引起科研者的广泛关注,该通路参与了HIV-1 感染引起的巨噬细胞凋亡、寿命延长;影响了巨噬细胞的自噬作用;介导了巨噬细胞、CD4+T 细胞的病毒储存库的产生;神经胶质细胞的炎症反应等重要生理病理改变,并被广泛研究,且该通路也作为治疗HIV-1 的重要靶标被深入研究。
虽然针对HIV-1 相关的PI3K/AKT 通路的研究已经在如火如荼地进行中,但针对PI3K/AKT 通路的抗HIV-1 药物还有极大的研究空间,尤其在针对PI3K/AKT 信号通路的抗HIV-1 药物的研究上,和抗肿瘤等药物一样在研发上需要投入更多精力。PI3K/AKT抑制剂在抗肿瘤方面取得了一定的成果,虽然在临床应用上因为药物活性减低、患者耐受力下降和出现皮疹和腹泻等副作用而终止,但仍有抗肿瘤PI3K/AKT 抑制剂通过临床试验后上市,如艾德拉斯司、阿培利司[42]。AIDS是一种复杂且极其难治愈的疾病,若想将靶向PI3K/AKT 通路的药物应用于抗AIDS上仍需要在其基础机制、药物筛选和临床试验等方面做出巨大努力。
PI3K/AKT 通路在对HIV-1 感染的机制研究上有很深的研究前景,如HIV-1 感染巨噬细胞既能引起靶细胞的自噬、凋亡,又能引起巨噬细胞的寿命延长。区别于巨噬细胞的寿命延长,CD4+T 细胞在HIV-1 感染后迅速地耗竭是否也与该通路有关系?是否有可能筛选出针对该通路抗HIV-1感染引起的CD4+T 细胞损伤的药物?诸如此类的该通路关于HIV-1感染的机制研究还有待进一步探索。
猜你喜欢
九江学院学报(自然科学版)(2022年2期)2022-07-02
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
新世纪智能(数学备考)(2021年10期)2021-12-21
昆明医科大学学报(2021年10期)2021-12-02
天津医科大学学报(2021年4期)2021-08-21
现代临床医学(2021年2期)2021-03-29
分析化学(2017年12期)2017-12-25
安徽医科大学学报(2015年9期)2015-12-16
安徽医科大学学报(2015年9期)2015-12-16
