
面向大规模人群的基因组注释系统
2022-04-01 08:56闫贞磊国宏哲
生物信息学 2022年1期
闫贞磊,国宏哲
(哈尔滨工业大学 计算学部,哈尔滨 150001)
1 材料与方法
1.1 数据下载
注释数据来源于多个公共数据库,下载的数据经过格式及参考基因组版本转换以备注释使用。
1.1.1 Annovar整合数据下载
Annovar数据下载方式为:annotate_variation.pl -buildver hg38 -downdb -webfrom annovar dataname humandb/。从公共数据库annovar[1]下载了包含人群变异频率数据、疾病变异数据、祖源数据、SNP数据、全基因组关联分析数据、保守性数据、变异位置及功能等数据。
1.1.2 CADD数据下载
CADD数据需要下载多版本数据,目前使用的是1.6(hg38/hg19),注意下载的数据是拆分了snv和indel的,都要下载。
从公共数据库CADD[2]下载了包含bscores、chromhmm、dbscSNV、encode、encodeMotifDB、exons、gerp、grantham、mirSVR、mirTargetScan_v71、mmsplice、mutation_density、phastCons、phyloP、reference、regulatoryBuilt、Remap、spliceai、transcripts、vep等数据。
1.1.3 UCSC数据库数据下载
UCSC数据库使用FTP下载方式在HG38 goldpath路径下载数据,同时也通过Tablebrowser下载数据。
从UCSC[3]数据库下载的数据分为phastCons20way placental、phastCons100way vertebrate、phyloP20way placental、phyloP100way vertebrate、Recombinate Rate、repeatmasker、TAD等。
1.1.4 Fantom 5数据库
CAGE Promoters和CAGE Enhancers 下载于Fantom 5[4]数据库,直接选择hg38版本的F5.hg38.enhancers.bed.gz、hg38_fair+new_CAGE_peaks_phase1and2.bed.gz文件。
1.1.5 Ensembl Regulatory数据
Ensembl Regulatory Build 下载于Ensembl[5]数据库,提供的链接可能直接复制下载会失效,可以按照链接提供的目录自己逐级查找,下载的文件为homo_sapiens.GRCh38.Regulatory_Build.regulatory_features.20190329.gff.
1.1.6 ORegAnno3 下载(2016)
ORegAnno3[6]存储为调控元件信息数据库,数据处理后保留所有位置信息及调控原件信息内容。
1.1.7 LINSIGHT得分
LINSIGHT[7]是一个统计模型,用于估计人类基因组中非编码序列的阴性选择。LINSIGHT得分衡量了非编码位点上的阴性选择概率,可用于对与遗传疾病相关的SNVs进行优先排序,或量化调控序列(如增强子或启动子)的进化约束。
1.1.8 miRNA数据
miRNA信息来源于miRBase r21[8]and snoRNABase v3[9]。
1.1.9 GenCode 数据
GenCode[10]数据包含基因位置、名称、转录本、类型等信息。
1.2 数据格式及参考基因组版本转换
所有已经下载的数据参考基因组版本为hg38的数据不需要进行参考基因组版本转换,将其处理成注释可用格式即可。参考基因组为HG19的数据需要进行数据版本转换,版本及格式转换完成后的数据处理为varnote可用的知识库,以供varnote工具对变异数据进行注释。
1.2.1 liftover等工具进行版本转换
liftover的获取地址:wget http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/liftOver。同时,获取坐标注释文件hg38Tohg19和hg19Tohg38,且获取命令如下:
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg38/liftOver/hg38ToHg19.over.chain.gz
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/liftOver/hg19ToHg38.over.chain.gz (下载得到的*over.chain.gz不需要解压)其他版本的注释文件可在UCSC数据库查找。
Input文件 BED格式文件,BED格式文件只定义前三列:chr start end,无表头(注:start不等于end)。坐标转换命令:liftover inputfile,over.chain.gz,outputfile,unmapfile。
1.2.2 其他版本转换工具
部分文件转换版本的时候用的是CrossMap.py[11],尤其是hg18及更老版本转换到hg38的版本的时候,可以实现一步转换。CrossMap.py bed *_to_*.chain.gz input.bed out.bed。
1.2.3 bw文件处理
bw文件可以用于UCSC及IGV等可视化工具展示,但是注释的时候不适合使用,要转换问bed文件才能进行注释。bw文件转换使用的是UCSC提供的bigWigToBedGraph[12]工具,运行命令为:bigWigToBedGraph file.bw out.bedGraph(如果是HG19的数据按照上面版本转换方法进行版本转换)。
1.2.4 VCF数据预处理
所有.vcf格式变异均处理为avinput(参见annovar avinput处理)格式,知识库及得到的变异数据文件都要进行相应处理,vcf格式知识库数据还包括GWAS、GRASP和CADD等文件。
1.3 数据注释
1.3.1 流程结构(见图1)

图1 基因组注释流程图Fig.1 Flow chart of genome annotation
1.3.2 注释步骤
转换avinput:avinput文件是annovar的标准输入文件,其自带数据库均依据该文件中ref/alt的格式建立,当前注释流程的依赖数据库中包含了很多annovar自带的数据库,且无法通过avinput的ref/alt格式逆推回原本的格式。同时,avinput文件定义的ref/alt格式可以有效地将整合后的vcf文件中的ref/alt标准化,如原本的ref/alt为C/T,但整合后有可能会变为AC/AT,显然整合后的ref/alt并不能与数据库中的信息匹配,故标准化是保证注释命中的关键步骤。由于annovar自带的标准化过程限定条件太多,部分不满足要求的行会被自动过滤掉,且这一过程无法通过参数进行调节,故这一步骤采用了改写annovar代码的方式,自行编写出了python版本的转换代码。
拆分同位置上多allele:由于该注释流程获得的上游输入文件是多个样本整合后的vcf,而整个项目最终会需要将多次收到的输入文件进行整体整合。对于单个收到的vcf来说,其中样本的基因型是依照当前的vcf给出的,这一基因型会与最终整合的vcf不匹配。同时,转换后的avinput每一个allele都是单独一行,若不进行拆分则不利于后续的整合步骤。
1.3.3 annovar注释refGene
refGene是基于基因的注释,varnote无法注释该类型的数据,故采用annovar对该数据库进行注释。
1.3.4 varnote注释
生成varnote配置文件:基于注释框架中指定的注释库、项目、输出项目名自动化生成varnote使用的配置文件。
通过avinput建立数据库:varnote进行注释时,对于完全注释不到的行,不会在结果文件中进行输出,这样的输出结果不符合实际需求以及后续整合的需要,且这一行为无法更改。故将转换后的avinput文件建立为一个数据库,通过自己注释自己的方式,使得每一行信息都能得到注释,从而保证结果文件中不会丢行。
注释:通过读取流程框架中配置文件的数据库信息、参数信息拼接出varnote的实际执行语句进行调用注释。
1.4 整合注释结果
1.4.1 去除无用数据列
无用数据列包括原始vcf文件中包含的部分信息、avinput数据库产生的额外信息等。在最后的整合过程中予以剔除。
1.4.2 重新对数据列排序
根据配置文件中指定的数据列排列顺序,将注释结果的信息列进行重新排序。
1.4.3 整合refGene注释结果
将annovar单独注释出的refGene信息整合到最终结果文件中。
1.4.4 整合样本基因型
将拆分后的多allele样本基因型合并到注释结果中,形成最终结果文件。
1.5 AnnotSV注释
1.5.1 注释各类型SV变异
结构变异注释的输入文件是根据SV类型产生的多个vcf文件,部分类型的文件可能不存在或内容为空。注释时给定的输入文件参数并非是实际的文件名,而是前缀名,经过拼接后自动查找目录中是否有相应类型的文件,逐个调用annotSV进行注释。
1.5.2 整合SV注释结果
由于输入文件多个文件,产生的结果文件同样是多个,且无法保证每种SV类型的结果文件都存在。对各个类型的结果文件进行检验,查看其是否存在,将存在的文件进行合并拼接,得到最后的完整SV注释结果。
2 结果与分析
2.1 注释数据统计
2.1.1 人群变异频率数据
为了进行注释收集整理了多类变异频率数据,其中包含人群变异频率数据,例如:the Exome Aggregation Consortium(外显子组整合数据库)、Genome Aggregation Database[13](简称gnomAD)是由各国研究者联合发展起来的基因组突变频率数据库,汇集众多大规模测序计划的基因组数据、NHLBI 计划汇集的外显子组突变频率数据、1000G 计划汇集的全基因组突变频率数据、KAVIAR[14]整合了HGP、1000G、CGI69、UK10K等共35个计划的基因突变频率数据、HRCR整合了32 k个样本整合的基因组突变频率数据、巴西人群外显子组突变频率数据[15]、中东地区人群外显子组变异频率数据、以及收集整理的中国人群变异频率数据。我们对人群变异频率数据的总数据量、SNP/INDEL数量、稀有突变、低频突变、高频突变的数量进行了统计,各人群数据库内稀有变异比例最高,SNP数量大于INDEL数据量,位点数量最多的数据库为gnomad,近3亿个变异位点,其他全基因组数据库包含变异位点数量大多小于1亿个。统计结果(见图2)。
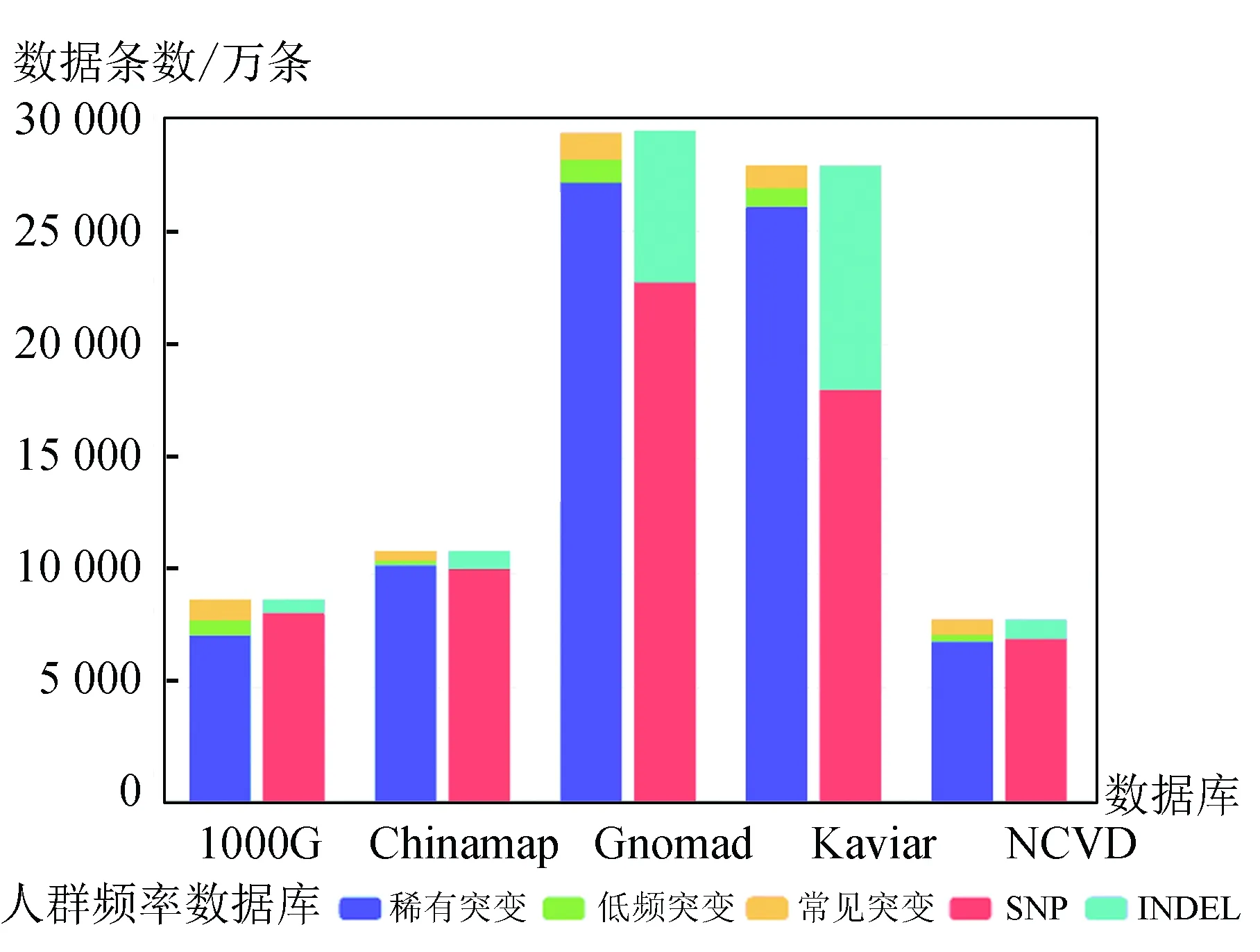
图2 人群变异频率数据Fig.2 Illustration of frequency of population genome variation
2.1.2 临床及表型注释数据
为了了解变异是否是已知的致病变异我们整理了多个DNA变异和人类疾病关系知识库,包括:60种人类肿瘤细胞系外显子组变异频率数据、预测内含子单核苷酸变异的致病性影响数据库、NCBI维护的疾病相关的人类基因组变异数据库、EBI负责维护的一个收集已发表的GWAS研究的数据库[16]、GRASP 2.0[17](SNP和表型关联数据库)、癌症基因突变数据库[18],此外还有基于美国医学遗传学与基因组学学会(The American College of Medical Genetics and Genomics,ACMG)曾制定过序列变异解读指南而分析变异数据的数据库-InterVar[19]。我们统计了这些疾病及表型相关的数据库内的变异数量及分类情况,大多数变异数据和临床关系 为“未明确”或者“可能致病/良性”,明确的“良性”及“致病性”位点比例较低,InterVar和clinvar统计结果(见图3和图4)。
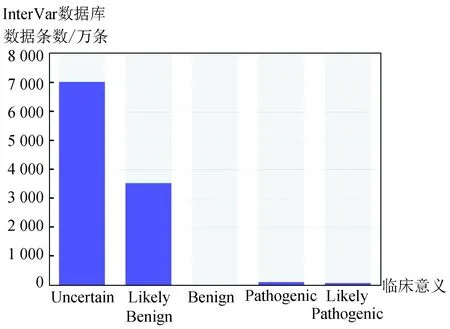
图3 InterVar临床变异数据统计Fig.3 Statistics of clinical variation based on the InterVar database
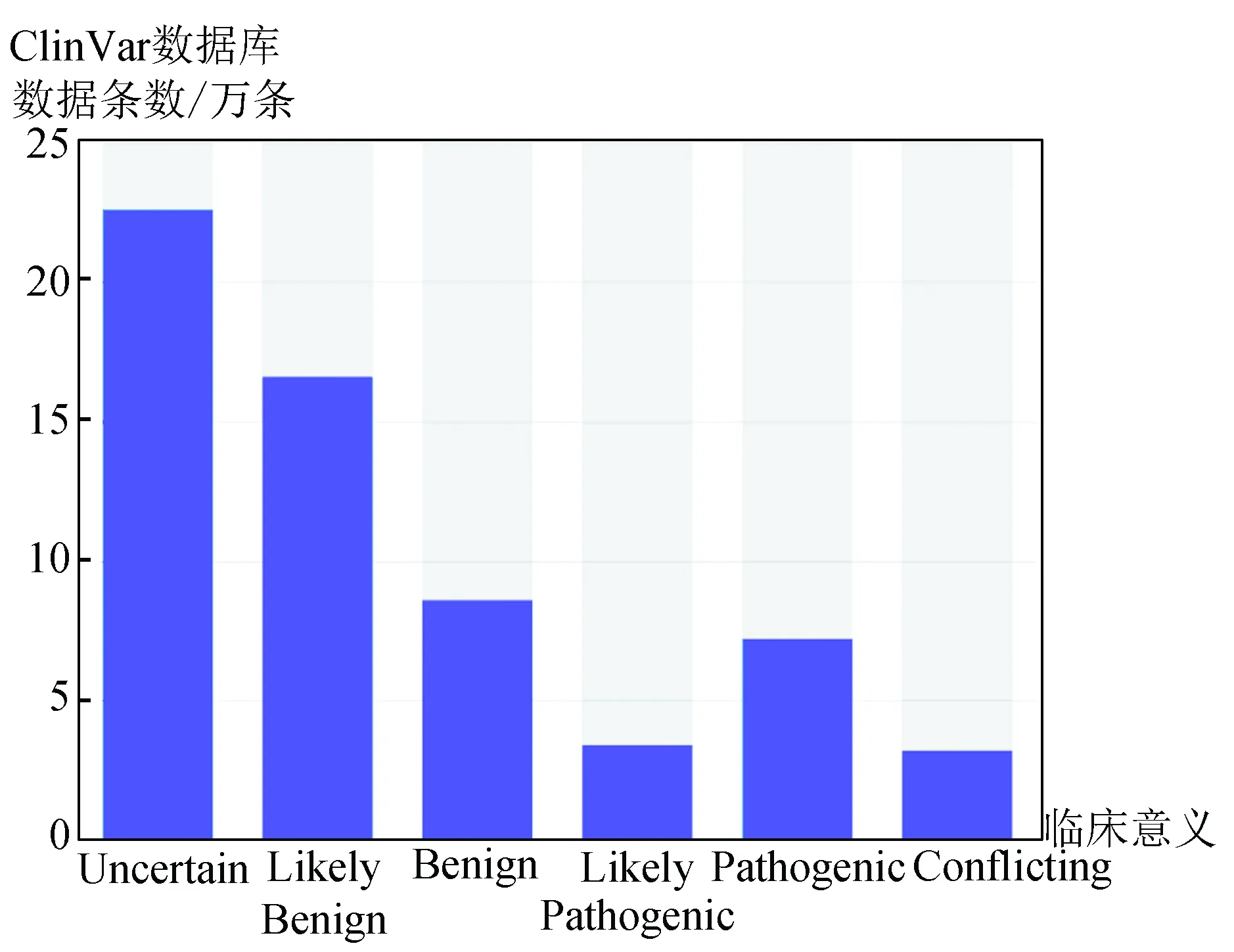
图4 Clinvar临床变异数据统计Fig.4 Statistics of clinical variation based on the clinvar database
2.1.3 序列保守性数据库
收集整理了多个评估序列保守性的数据库以及对相应保守性的得分信息,比如灵长类动物基因组序列PhyloP[20]方法保守性打分数据、哺乳动物基因组序列PhyloP方法保守性打分数据、脊椎动物基因组序列PhyloP方法保守性打分数据、灵长类动物基因组序列PhastCons方法保守性打分数据、哺乳动物基因组序列PhastCons方法保守性打分数据、脊椎动物基因组序列PhastCons方法保守性打分数据、由GERP++[21]方法计算得到的GerpN值等,同时还下载整理了祖源等位基因情况。以上的保守性打分数据对基因组上所有的位点进行打分,每个位点都有相应的分值。
2.1.4 表观注释数据
为了对变异区域进行表观注释,我们下载了多个相关的数据库,其中包括识别promoter和enhancer区域的数据、细胞中染色质标记状态比例数据、预测得到的转录因子motif数量的数据、与转录起始和终止位置的距离数据、表观遗传学标记和检测相应转录调控原件的数据、染色质拓扑结构域识别的数据等。
2.1.5 变异综合得分
为确定每个变异的功能和致病性风险程度,多个数据库基于多种类的注释结果对变异数据进行了综合打分,这些数据库包括了LINSIGHT得分,LINSIGHT用于估计人类基因组中非编码序列的阴性选择。LINSIGHT得分衡量了非编码位点上的阴性选择概率,可用于对与遗传疾病相关的SNVs进行优先排序,或量化调控序列(如增强子或启动子)的进化约束。还包含了在编码区和非编码区对遗传变异进行注释的Eigen[22]得分、CADD得分、fitcons[23]得分等。各数据库包含位点数量(见表1)。

表1 变异综合得分总数Table 1 Total number of variants from multiple databases
2.1.6 变异基础信息
变异区域的GC比例,以及变异所在的区域所在的功能区,变异在相应的功能区域的相对位置,例如是否位于promoter区域,是否在编码区域,在第几个外显子上,在第几个内含子上,这些变异的信息相关的数据都进行了整理,以及用dbSNP(150版本)数据对每个位置的变异进行了注释。这些信息覆盖整个基因组。
2.1.7 蛋白功能数据
基因变异会对蛋白功能造成相应改变,基于氨基酸序列的同源性和物理性质来预测氨基酸的替换对蛋白质功能是否造成影响,用来评估基因变异的有害程度的数据有SIFT[26]得分及polyphen[27]得分,除了这两种打分数据我们还整理了MA[28]、LRT[29]等蛋白功能预测数据对基因组变异进行打分。
2.1.8 miRNA数据
MicroRNA (miRNA) 是一类内生的、长度约为20-24个核苷酸的小RNA,其在细胞内具有多种重要的调节作用。每个miRNA可以有多个靶基因,而几个miRNA也可以调节同一个基因。miRNA的基本信息及靶向关系的信息来源于miRBase和TragetScan[30]数据库等数据库,各库包含的数据量(见表2)。

表2 miRNA数据量统计Table 2 Statistics of miRNA based on multiple databases
2.1.9 结构变异注释数据
结构变异注释使用的注释工具室AnnotSV[31],注释数据使用的是软件自带的,注释项目包括:变异的基本信息,比如变异所在的转录本,所在的CDS长度,变异长度等信息,还提供变异位置其他人群机构变异频率的注释,比如DGV、gnomAD、1000G、IMH数据库。变异致病性注释使用的是dbVAR、ClinGen、clinvar等数据。结合以上所有信息对相应的结构变异进行打分,得出致病性结论。
2.2 注释流程涉及软件及工具
2.2.1 LiftOver
将所有的数据转换为同一个参考基因组之后进行注释是一个必经的步骤,LiftOver是一个高效的位置信息转换工具,在我们使用工具进行转换的时候,成功转换的比例大于99.97%,2 000万条变异的数据进行基因组位置转换在五分钟内完成,注释消耗时间与目标文件的大小成正比。
2.2.2 Annovar
Annovar[1]是一个高效的软件工具,可以利用最新的信息对从不同基因组(包括人类基因组hg18、hg19、hg38,以及小鼠、蠕虫、苍蝇,酵母和许多其他基因组)检测到的遗传变异进行功能注释。给出带有染色体、起始位置、结束位置、参考核苷酸和观察到的核苷酸的变异列表。其功能包含基于基因的注释、基于区域的注释、基于过滤的注释及其它辅助功能。
在工作流中,Annovar负责对样本VCF进行基于基因注释,主要依赖于refGene数据库。在测试过程中,针对500万行的VCF文件,完成注释工作需要耗时约8分钟,针对2 500万行的VCF文件,完成注释工作需要耗时约40分钟。可见ANNOVAR的工作效率是比较稳定的,注释消耗时间与目标文件的大小成正比。
2.2.3 VARNOTE
VARNOTE[32]是ANNOVAR的作者最新开发的注释工具,该工具主要针对基于过滤及基于区域的注释,由于算法上的优化,其注释效率较ANNOVAR有极大的提升,且注释过程中不再需要将依赖的数据库完整读入内存中,有效地降低了对机群资源的占用。使用VARNOTE进行注释需要自行构建依赖的数据库,这一过程由于需要进行排序、压缩、建立索引等过程,相较于ANNOVAR比较繁琐,但VARNOTE的效率提升主要便是依托于对依赖数据库的处理,故这一过程必不可少。
在工作流中,VARNOTE负责对样本VCF进行所有的基于过滤以及区域的注释工作,当前包含的数据库共56个。在测试过程中,针对500万行的无INDEL位点VCF文件,VARNOTE对其进行56个数据库的注释仅需要3分钟。但在实际工作过程中,VARNOTE的注释效率波动较大。由于其算法原因,VARNOTE对INDEL位点的注释效率较低,当目标VCF文件中INDEL位点的占比较大时,其注释效率会有比较明显的下降。但整体而言,VARNOTE的注释效率仍然是比较理想的。
2.2.4 AnnotSV
AnnotSV是一款使用Tcl语言编写的用于注释以及评估结构变异的软件,其目的在于发现结构变异的潜在致病性以及过滤出假阳性结构变异。该软件接受VCF格式以及BED格式的文件作为输入,以tab分割的文本文档作为输出文件。AnnotSV在注释时需要使用各种不同的参考数据库来进行支持,这部分不需要用户自行处理,在注释过程中若本地不存在相应的数据库,软件会自动进行下载操作。
在工作流中,AnnotSV负责对结构变异数据进行注释工作。在测试过程中,针对27 000行的结构变异VCF文件,完成注释工作需要13分钟,针对49 000行的结构变异VCF文件,完成注释工作需要25分钟。注释消耗时间与目标文件的大小大体上符合线性规律。
2.3 变异注释结果统计
对注释后的样例数据结果进行统计,发现在示例样本中变异类型中大多数是单碱基替换变异,插入和缺失变异数量相近,并且远少于单碱基替换位点数量,样例数据中位点变异数量超2 000万个。多数变异位点位于基因间区及内含子区域。因为样例数据中单碱基替换位点比例较高,蛋白结构变异类型中同义突变及非同义突变数量最多,非同义突变数量大于同义突变。样例数据涉及变异类型统计结果(见图5),样例数据变异位点区域分布(见图6)。
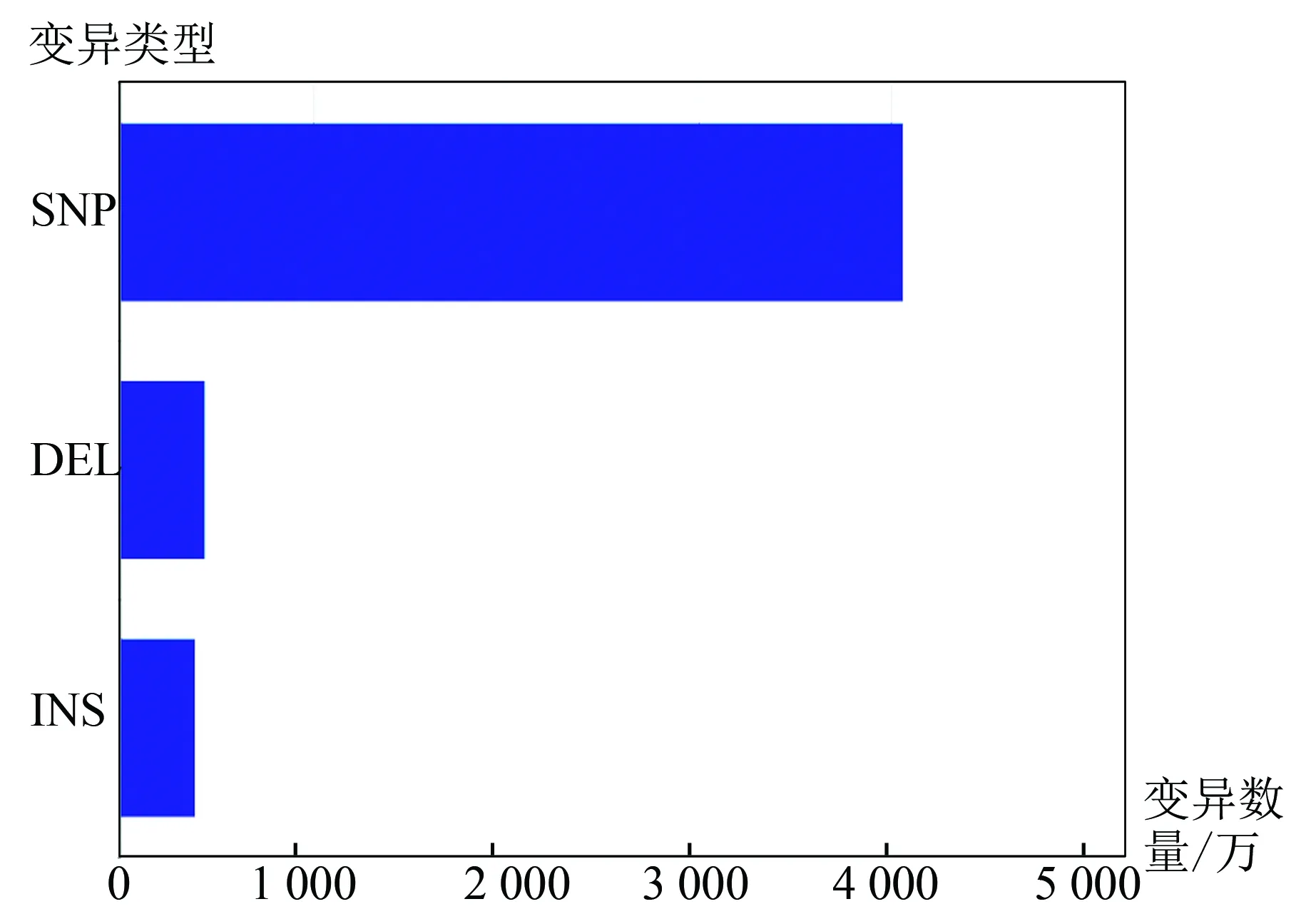
图5 样例数据变异类型数量Fig.5 Statistics of multiple types of variation
因为样例数据中单碱基替换位点比例较高,蛋白结构变异类型中同义突变及非同义突变数量最多,远超移码突变,非同义突变数量大于同义突变,而且占总突变数量的一半以上。移码突变中,不是3或3的倍数碱基缺失或增加的移码突变(编码氨基酸序列改变)数量少于其他移码突变数量。突变导致终止密码子获得或缺失在所有变异类型内比例最低。编码区蛋白结构变异类型比例(见图7)。
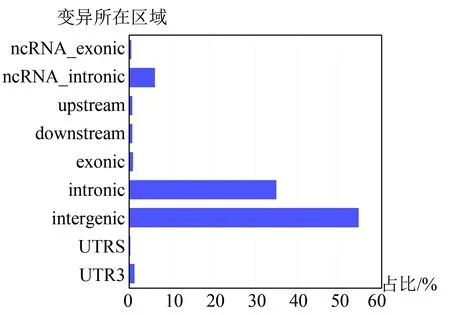
图6 样例数据变异所在区域分布比例Fig.6 Statistics of regional distribution of genomic variation
2.3.1 转换和颠换
碱基颠换(transversion)是指在碱基置换中嘌呤与嘧啶之间的替代,而转换(transition)则是一个嘌呤被另一个嘌呤,或者是一个嘧啶被另一个嘧啶替代。在样例数据中,转换变异数量是颠换变异数量两倍有余(约2.1倍),其中G>T和C>A碱基变异比例最高,均超过了全部变异数量的20%,T>G和A>C次之,均超过全部变异数量的10%,各种颠换变异的碱基比例均较低。各碱基变异情况统计结果(见图8)。

图7 样例数据编码区蛋白结构变异类型比例Fig.7 Statistics of ratio of protein structural variationin encoding region
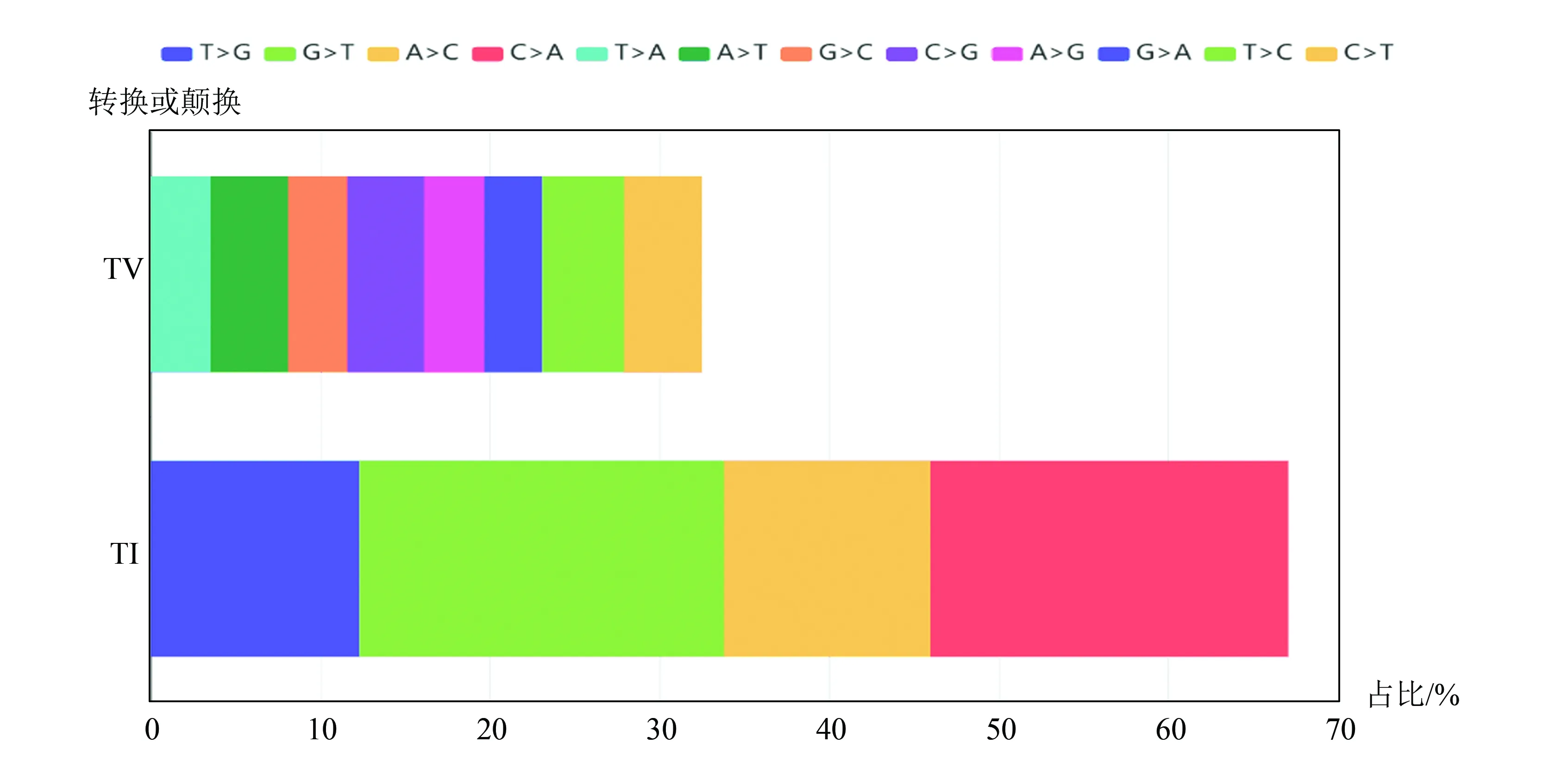
图8 转换及颠换变异比例Fig.8 Statistics of ratio of transition and transversion variations
2.3.2 样本变异数量分布
样例数据中每个样本的变异数量均有所差异,经过我们统计单样本变异数量在470万到500万区间,样本变异数量在485万附近的样本最多,样本变异数量分布(见图9)。
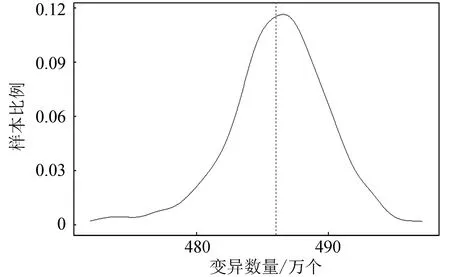
图9 样本变异数量分布Fig.9 Distribution of variation quantity
2.3.3 结构变异数量分布
每个样本中有多种类型的结构变异,样例数据的结构变异数量在7 000到1.2万之间,大部分样本内的结构变异数量在9 000个左右,样例数据结构变异数量分布(见图10)。
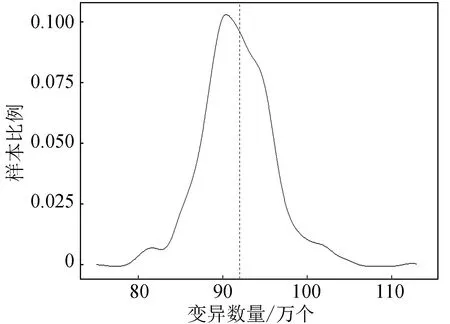
图10 样本结构变异数量分布Fig.10 Distribution of the number of structural variations
3 结 论
1)基因组注释是确定基因组序列中基因及编码区位置的过程,其可以直接对序列赋予生物学意义,故基因组注释是解读基因组序列重要途径。基因表达及变异对其生物学功能发挥具有决定性意义,通过基因组注释,可以确定基因调控区域及编码区域是否变异,以及变异的影响,从而帮助研究者从生物功能的角度理解基因组。个体表型的差异究其根本是由于遗传物质的差异造成的,通过基因组注释可以帮助研究者发现个体差异的遗传本质,发现群体的遗传特征。此外,通过对基因组进行注释,结合已有变异与表型的关联知识,可以评估个体的健康情况并指导个性化用药。
2)我们研究开发的面向大规模人群的自动化基因组注释系统利用国际具有高置信度的人类基因组变异数据库,综合运用不同注释方法和高效的索引技术,实现了对个人基因组和群体基因组的高效、快速,准确和全面的基因组信息注释。注释系统对每一个变异位点实现了基因组区域类型、变异类型、基因类型等基础基因组相关信息标注。同时,系统对每一个变异位点进行综合功能性分析,例如:基因组转录、甲基化、功能损失以及各类疾病的风险预测等分析。该注释系统计算并展示个人基因组的差异化特征,助力预测个体基因组变异的相关疾病的发病风险。同时,该系统可应用大规模人群基因组数据分析中,提供全面且准确的基因组变异数据的注释服务,为不同地区和民族的人群遗传学分析提供了坚实基础。
猜你喜欢
今日农业(2021年11期)2021-08-13
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
趣味(数学)(2020年4期)2020-07-27
支部建设(2020年15期)2020-07-08
广东教学报·教育综合(2020年15期)2020-03-23
中成药(2018年7期)2018-08-04
知识文库(2017年9期)2017-10-20
百科知识(2015年18期)2015-09-10
中学教学参考·文综版(2014年1期)2014-03-11
