
Theoretical study on the mechanism for the excited-state double proton transfer process of an asymmetric Schiff base ligand
2022-04-12 03:45ZhengranWang王正然QiaoZhou周悄BifaCao曹必发BoLi栗博LixiaZhu朱丽霞XingleiZhang张星蕾HangYin尹航andYingShi石英
Chinese Physics B 2022年4期
关键词:石英
Zhengran Wang(王正然), Qiao Zhou(周悄), Bifa Cao(曹必发), Bo Li(栗博), Lixia Zhu(朱丽霞),Xinglei Zhang(张星蕾), Hang Yin(尹航), and Ying Shi(石英)
Institute of Atomic and Molecular Physics,Jilin University,Changchun 130012,China
Keywords: DFT/TDDFT,schiff base ligand,excited state intramolecular double proton transfer
1. Introduction
Photoexcited proton transfer has been investigated intensively both experimentally and theoretically,because of its important role in physical and biological systems.[1-3]The first discovered excited state proton transfer phenomenon was discovered by Weller and co-workers in the characteristic experiment of methyl salicylate.[4]Molecules with excited state intramolecular proton transfer(ESIPT)properties could exhibit dual fluorescence peaks under specific photo-excitation.[5,6]One of the fluorescence peaks could derive from the initial molecular structure before ESIPT behavior (enol configuration),and the other one results from the form after ESIPT reaction(keto structure).[7]In general,most ESIPT reactions are involved with single proton transfer. However, such a single proton transfer could not be sufficient to simulate the proton transfer process thoroughly in the biological field,because the most photoinduced mutation of DNA or RNA refers to double or multiple protons participating.[8-12]Tayloret al.found that the 7-azaindole dimer has a different tautomer in the ground and first excited states,which is the first recorded example of concerted double proton transfer.[13]Since then, the excited state double proton transfer(ESDPT)process has been extensively investigated due to its potential applications in chemical reactions and alkaloid base pair variation.[14-17]
Schiff base ligands with proton transfer properties have been studied extensively due to their thermal,photochromatic,complex properties and tautomeric equilibrium.[18-22]They are classified as symmetric and asymmetric ligand based on contracture. Asymmetric Schiff base ligands can provide appropriate donor sets capable of holding three metal ions in a linear complex. Recently, asymmetric Schiff base ligands have been attracting considerable attraction, which are very useful ligands in the synthesis of transition metal complexes.[23-28]Samiraet al.synthesized 1-[(2-hydroxy-3-methoxy-benzylidene)-hydrazonomethyl]-naphthalen-2-
ol(HYDRAVH2) ligand.[29]THYDRAVH2is an asymmetric fluorescence Schiff base ligand with the properties of ESDPT and remarkable optical properties. In Samiraet al.’s study, the structure of the HYDRAVH2was characterized.And the experimental results indicate that the HYDRAVH2ligand displays particularly enhanced emission properties in near-infrared,and has great application value in optoelectronic devices. It can be used to synthesize a series of new fluorescent lanthanide (III) complexes which caused an increase in fluorescence.[30]Due to the asymmetry of its structure, it has a certain impact on the double proton transfer. However, the mechanism of the ESDPT process in the HYDRAVH2system is still unclear.
In the present work, we optimized the structures of the HYDRAVH2 in S0state and S1state by the density functional theory(DFT)method and the time-dependent DFT(TDDFT)method,respectively.Moreover,the non-covalent interactions,frontier molecular orbitals and potential energy surfaces were presented to explore the double proton transfer mechanism.
2. Theoretical calculation methods
In this work, the related calculations were completed on the Gaussian 09 program suit.[31]The ground- and excitedstate geometric configurations of the HYDRAVH2monomer in enol and keto forms were optimized in the framework of DFT and TD-DFT methods, respectively, with the B3LYP functional and 6-311+G(d) basis set.[32,33]The 1H NMR spectra of structure was done on the optimized geometries at the same computational level. The 1H NMR chemical shifts of the HYDRAVH2were predicted with respect to the tetramethyl silane (TMS) by employing the gaugeindependent atomic orbital method.[34-36]In view of the acetonitrile (ACN) solvent adopted in previous experiment, the ACN solvent with integral equation formalism variant of the polarizable continuum model(IEFPCM)was used in theoretical arithmetic.[37,38]Based on the optimized configurations,the pivotal bond lengths, bond angles, frontier molecular orbitals, absorption and emission properties, and infrared (IR)vibration spectra were calculated.[39]The reduced density gradient(RDG)images were drawn by Multiwfn software.[40-42]In view of adopting potential energy curves to illustrate the ESDPT mechanisms, the S0and S1potential energy curves have been scanned by increasing the bond length of O22-H34and O11-H39in steps of 0.1 °A.H34,N14···H34,O11-H39,and N13···H39are 0.98 °A,1.78 °A,0.99 °A, and 1.72 °A, respectively. When the HYDRAVH2is photoexcited to S1state,these bond lengths change to 0.99 °A,1.73 °A,1.00 °A,and 1.69 °A,respectively. It is found that compared with S0state,the bond lengths of O22-H34and O11-H39increased in S1state,while the bond lengths of N14···H34and N13···H39shortened in S1state. Due to the asymmetry of the HYDRAVH2structure, the change value of the bond length of the two hydrogen bonds that can promote proton transfer is different.That is to say,the proton transfer abilities of H34and H39are contrasting.
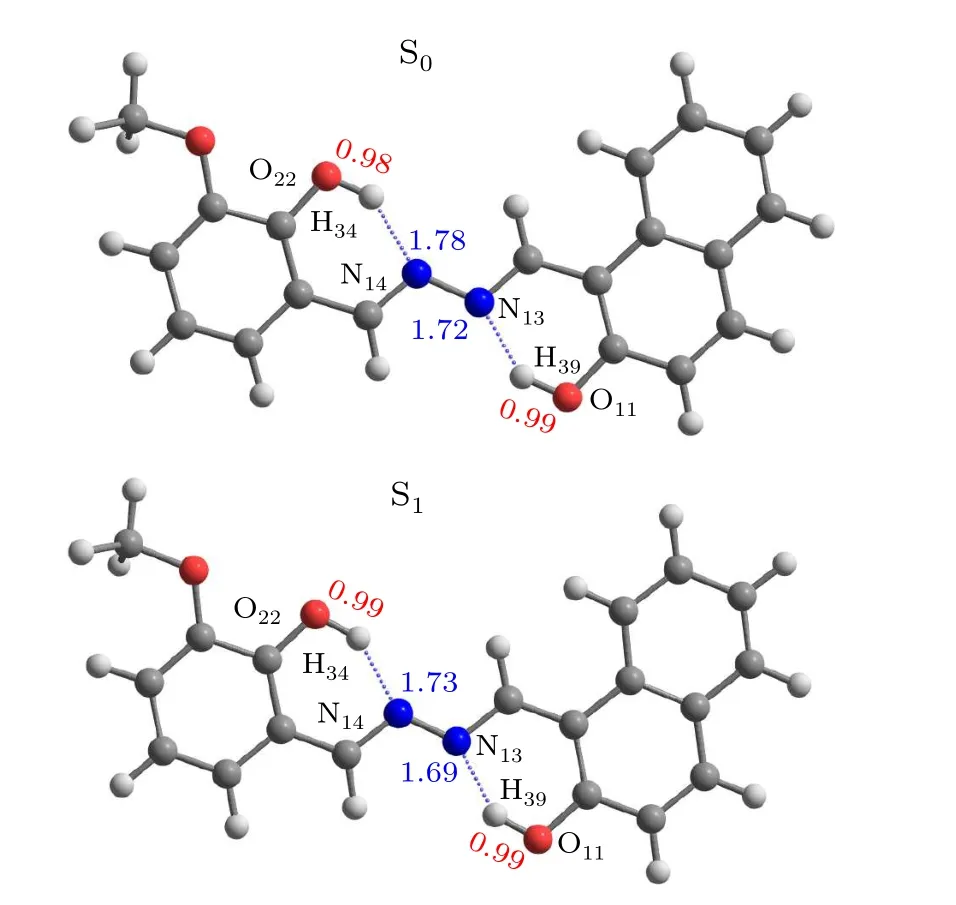
Fig.1. The optimized ground and excited states structures of HYDRAVH2.
3. Results and discussion
3.1. Optimized geometric structures
The ground- and excited-state geometric configurations of the HYDRAVH2monomer in enol forms are optimized as shown in Fig.1. The bond lengths(O-H and H-N)and bond anglesδ(O-H···N)related to the hydrogen bond are summarized in Table 1. In the S0state, the bond lengths of O22-

Table 1. Obtained bond lengths(°A)and bond angles(°)of the HYDRAVH2 in the S0 and S1 states,respectively.

Table 2. Chemical shift values(ppm)in 1H NMR spectra of HYDRAVH2 determined experimentally and theoretically by the B3LYP/6-311+G(d)level and IEFPCM model. The calculated chemical shift data for HYDRAVH2.
3.2. The 1H NMR spectra
The NMR spectroscopy is a powerful tool for the direct detection of chemical species and forms of molecular interactions in complex materials.[43]The 1H NMR spectra calculation of HYDRAVH2is shown in Table 2, which quite agreed with experimental NMR spectra.[29]The calculated isotropic shielding tensor for TMS is equal to 31.94 ppm for the 1H nuclei.[44]The chemical shifts of all involved protons are listed in Table 2. Similarly, the calculated proton chemical shifts of H39 and H34 are arrived at 11.459 ppm and 10.478 ppm,respectively. The calculated NMR chemical shift values are in good agreement with the experimental values. The results confirm the reliability of the calculated method we have chosen. In addition, it is worth noting that for all protons of HYDRAVH2, H34 and H39 shift the most considerably, suggesting there are two strong hydrogen bond interactions in OH···N.And the strength of the two hydrogen bonds is distinct.
3.3. Non-covalent interaction analysis

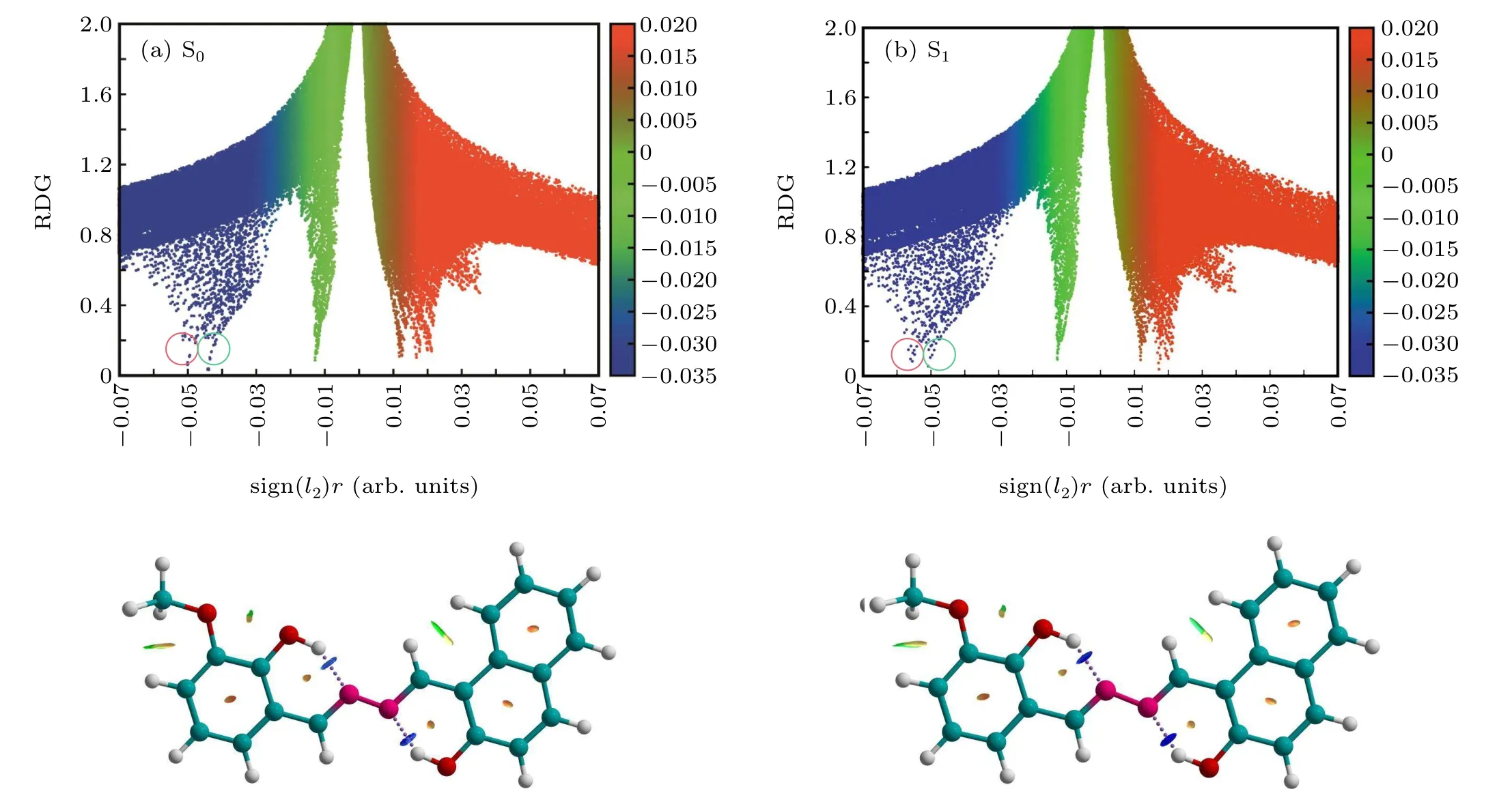
Fig.2. Plots of function value 1 versus function value 2 for HYDRAVH2 in(a)S0 and(b)S1 states,as well as the assignments of each spike by gradient isosurfaces.
Yanget al.introduced a method for imagining noncovalent interactions in real space,[45]which could explore intramolecular interactions. The different types of interaction and the homologous intensities can be definitely shown by analyzing the electron densities (ρ(r)) and their RDG isosurfaces. Specifically,the expression is The eigenvalueλ2>0 acts for bonding interactions and theλ2<0 represents anti-bonding interactions. TheΩ(r) negative value stands for hydrogen bond interaction. In contrast,the positive value ofΩ(r) means the presence of the steric hindrance. A nearly zero value of signΩ(r) indicates weak interactions. The scatter plots for the RDG value versus theΩ(r) value of HYDRAVH2in the ground-state and excitedstate are shown in Fig.2. The colors from blue to red indicate theΩ(r)value from negative to positive. As shown in Fig.2,the two spike peaks of the HYDRAVH2are observed and located at-0.045 and-0.051,respectively,in the S0state. The two spike peaks are assigned to O11-H39···N13(-0.045)and O22-H34···N14(-0.051),respectively. Upon photoexcitation to the S1state,O11-H39···N13and O22-H34···N14both shift from-0.045 to-0.050 and from-0.051 to-0.056. The left shift proves that the strength of two hydrogen bonds interaction in the HYDRAVH2increases in S1state.It is obvious that the intensity of interactions O22-H34···N14is stronger than O11-H39···N13. It can be concluded that the double transfer may not be synchronous in the excited state.
3.4. Frontier molecular orbitals
The frontier molecular orbital can reflect the distribution of electron density before and after molecular excitation clearly.[47]Figure 3 shows the electron density distribution of the highest occupied molecular orbital (HOMO)and the lowest unoccupied molecular orbital(LUMO)for the HYDRAVH2in the enol and keto states. It can be found from Fig.3 that the HOMO and the LUMO are characterized by theπandπ∗features, respectively. Herein, we just concentrate on the charge redistribution around hydrogen bonding moieties, which can affect excited state hydrogen bonding dynamics seriously. The electronic densities around O22and O11on HOMO orbital decrease compared with those on LUMO orbital,while the electronic densities around N atoms increase instead. This has significant facilitate effects on moving the hydrogen protons to the proton acceptors. Therefore,hydrogen bonds are strengthened in the photoexcitation process. Crucially, we can see that the change in electron density around O22is more pronounced than around O11,indicating that more electrons are transferred. Due to the difference in charge distribution around the donor O22and O11, the two oxygen donors have different driving forces for proton transfer,which directly leads to the fact that H34and H39cannot be transferred simultaneously.
Furthermore, by comparing the frontier molecular orbitals of the three keto states, we find that the strong coupling effect between the HOMO and LUMO happens in the keto1 form. However, when the HYDRAVH2is in the keto2 or keto3 state,the coupling degree of charge between HOMO and LUMO orbitals is lower than that of keto1 obviously.
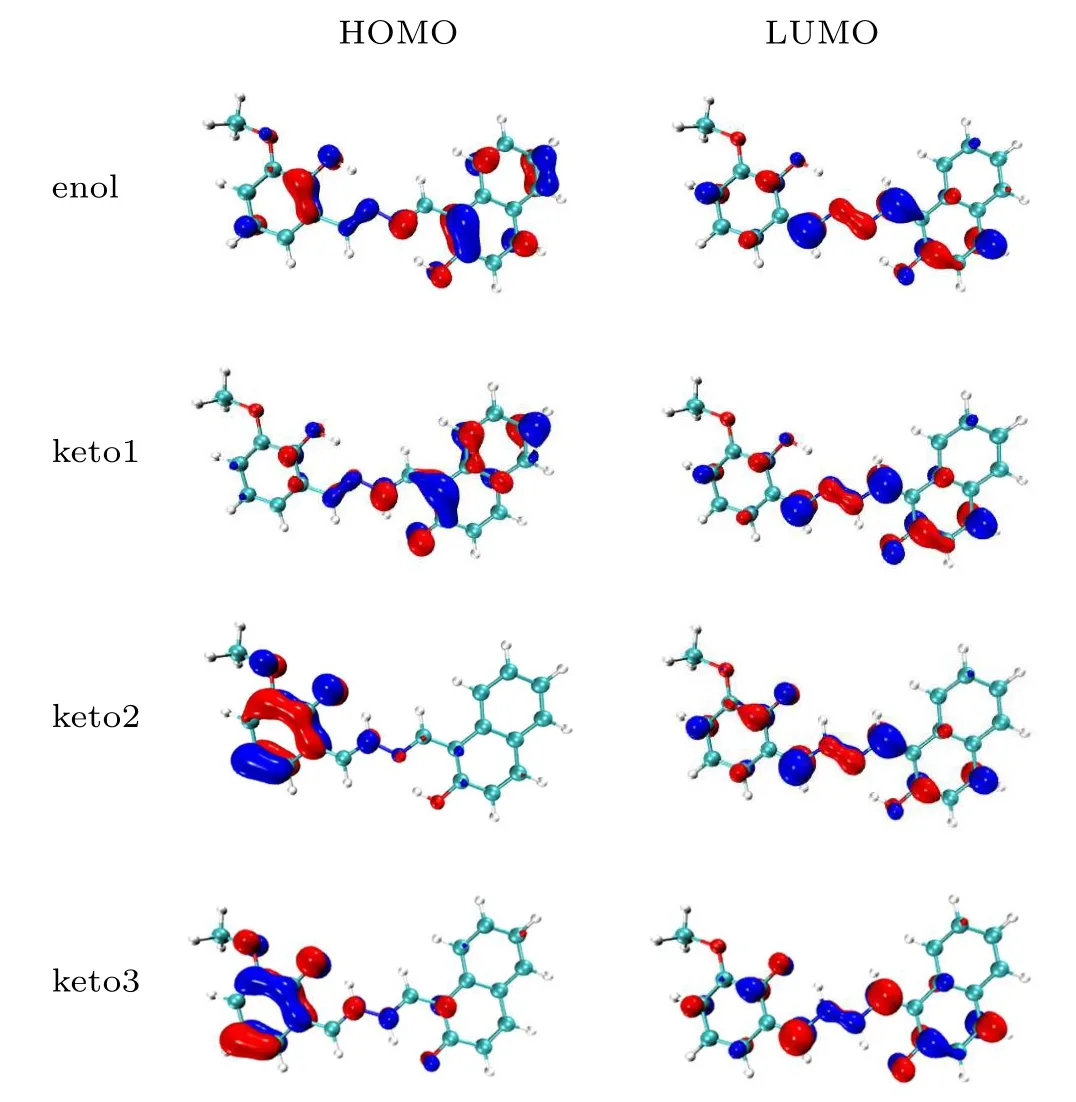
Fig. 3. The frontier molecular orbitals (HOMO and LUMO) of the HYDRAVH2 in the enol state and keto state.
3.5. Absorption and emission spectra
The theoretical absorption peak and emission spectra of HYDRAVH2in ACN solvent are calculated as shown in Fig.4.It can be seen from Fig. 4 that the absorption peaks of the HYDRAVH2are located at 338 nm and 397 nm, which stay closely aligned with the experimental results (332 nm and 387 nm).[30]We calculated two emission peaks and classified them as keto1 form and keto2 form, exhibiting the red shifts compared to the absorption peaks. The calculated emission peak of keto1 is 514 nm,which is in good agreement with the experimental values(495 nm).[30]In addition, we also calculated an emission peak of the keto2 form located at 664 nm.But in fact,only a single fluorescence emission was observed in the experiment. Combined with the frontier molecular orbital analysis above,the decouple effect between the electronic donor and the acceptor is a main factor for the nonfluorescent phenomenon.[48]Thus, we infer that the single fluorescence observed in the experiment should originate from the keto1 state,while the fluorescence cannot be emitted from the keto2 and keto3 states.
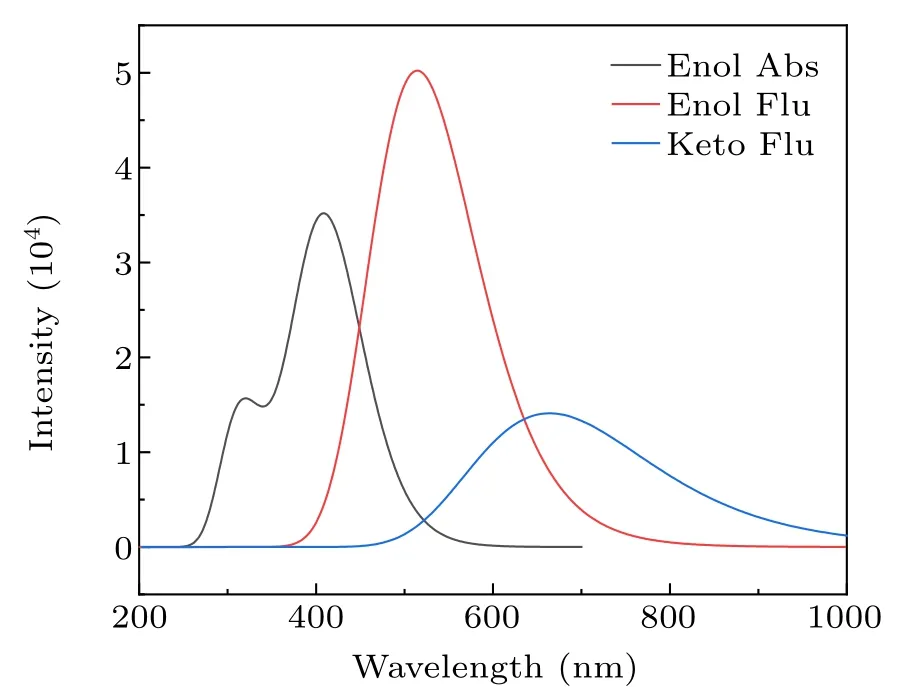
Fig. 4. The theoretical absorption peaks and calculated emission spectra of the HYDRAVH2.
3.6. IR vibration spectra
The calculation results of the IR spectra of the HYDRAVH2in the S0state and the S1state are shown in Fig.5. It can be seen that the O22-H34and O11-H39stretching peaks of HYDRAVH2in the S0state are located at 3276 cm-1and 3165 cm-1, exhibiting a red shift of 230 cm-1and 195 cm-1to 3046 cm-1and 2970 cm-1in the S1state, respectively. The red shift of the O-H stretching peaks for the HYDRAVH2suggests that the hydrogen bonds O-H···N are all strengthened in the S1state, promoting the occurrence of ESDPT.[49]Furthermore,it shows that in this state,the vibration frequency between O22-H34is stronger than that between O11-H39, indicating that the hydrogen bond of the former is stronger.

Fig.5. The calculated IR spectra of HYDRAVH2 in the S0 and S1.
3.7. Potential energy surfaces
In order to reveal the excited state intramolecular double proton transfer mechanism of the HYDRAVH2clearly,we constructed the potential energy surfaces of the HYDRAVH2in the S0state and the S1state.[50]The potential energy surfaces are scanned for molecule HYDRAVH2in S0and S1states with various bond lengths of O22-H34and O11-H39from 0.99 °A to 2.09 °A in steps of 0.1 °A and are shown in Fig.6.
As shown in Fig.6(a),in the ground state,the relationship of the four minima potential energy isEB=EC >ED >EA.PointsB,CandDin the figure correspond to keto1,keto2 and keto3 of the optimized configuration,respectively. The potential energy barriers among these points are calculated and presented as follows: 7.67 kcal/mol is needed to cross pointAto reach pointB(orC), the proton transfer from the enol (pointA) structure to the keto structure (pointD) needs to cross an energy barrier of 11.32 kcal/mol. Due to the high energy barrier of proton transfer,it is difficult for HYDRAVH2to spontaneously carry out the intermolecular double proton transfer process in the S0state.[51]
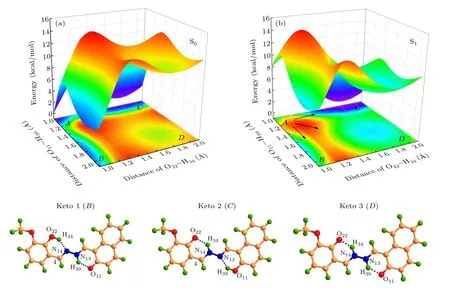
Fig.6. Potential energy surfaces of the(a)ground and(b)first excited states for HYDRAVH2 molecule along O22-H34 and O11-H39 bond lengths and the three possible configurations in the keto form.
Figure 6(b) shows the potential energy surface of the HYDRAVH2in the S1state. After excitation to the S1state,four structures denotedA,B,C, andDare observed. Among them,pointArepresents the enol structure of the HYDRAVH2,pointBand pointCrepresent the single proton transfer structure of the HYDRAVH2, pointDrepresents the keto structure of the HYDRAVH2after double proton transfer. The obvious decrease of energy barrier after photoexcitation indicates that the strengthening of the intramolecular hydrogen bond can promote the proton transfer. As shown in Fig.6(b),HYDRAVH2has the following three pathways to complete the excited state double proton transfer process,namely,A →B →D,A →C →D,andA →D. Among them,the first two paths are the stepwise proton transfer mechanism, and the third is the cooperative proton transfer mechanism. Specifically, the potential energy barriers among these points are calculated and presented as follows: the HYDRAVH2needs to cross the energy barrier of 4.65 kcal/mol from pointAto pointBand 2.43 kcal/mol from pointAto pointC. Then a higher barrier(6.43 kcal/mol) is required to reach theDpoint from theApoint. The two energy barriers are low enough for the proton transfer. However,it is difficult for the two protons to transfer simultaneously because of the higher barrier from pointAto pointD. It can be deduced that the double protons are transferred stepwise. Compared withA →Bprocess, the energy barrier needs to be crossed inA →Cprocess is lower,it shows that theA →Cprocess is more prone to proton transfer. These results identify that the HYDRAVH2can undergo ESDPT process in the first excited state and the transfer mechanism is a stepwise transfer mechanism.
4. Conclusion
Summing up,the excited state intramolecular double proton transfer of HYDRAVH2is theoretically investigated by DFT and TDDFT methods. The results of our investigation fully confirm that H34and H39have different proton transfer abilities by comparing the changes in the primary bond parameters. Frontier molecular orbital analysis shows that the increased charge densities on proton acceptor moiety could promote the ESDPT process. Furthermore,the IR vibration spectrum and the non-covalent interaction analysis further confirm the tendency of the ESDPT process. The results of potential energy surface analysis adequately prove the double proton transfer process is a stepwise proton transfer mechanism by comparing the heights of energy barriers. These distinctive characteristics provide valuable guidance for controlling the ESDPT process as well as the design and synthesis of new Schiff base ligands.
Acknowledgments
Project supported by the National Basic Research Program of China (Grant No. 2019YFA0307701), the National Natural Science Foundation of China (Grant No. 11874180),and the Young and Middle-aged Scientific and Technological Innovation leaders and Team Projects in Jilin Province,China(Grant No.20200301020RQ).
猜你喜欢
矿产保护与利用(2021年6期)2021-03-22
矿产勘查(2020年1期)2020-12-28
中国非金属矿工业导刊(2020年5期)2020-11-08
中国非金属矿工业导刊(2020年5期)2020-11-08
中国非金属矿工业导刊(2020年5期)2020-11-08
科学导报(2020年50期)2020-09-09
矿冶工程(2020年2期)2020-05-24
中国非金属矿工业导刊(2019年2期)2019-07-01
电子制作(2018年14期)2018-08-21
电子制作(2018年8期)2018-06-26

- Chinese Physics B的其它文章
- Quantum walk search algorithm for multi-objective searching with iteration auto-controlling on hypercube
- Protecting geometric quantum discord via partially collapsing measurements of two qubits in multiple bosonic reservoirs
- Manipulating vortices in F =2 Bose-Einstein condensates through magnetic field and spin-orbit coupling
- Beating standard quantum limit via two-axis magnetic susceptibility measurement
- Neural-mechanism-driven image block encryption algorithm incorporating a hyperchaotic system and cloud model
- Anti-function solution of uniaxial anisotropic Stoner-Wohlfarth model