
SHP2小分子抑制剂研究进展及构效关系分析
2022-04-15 03:03关兆辉徐学宇唐海徐云根
药学进展 2022年3期
关兆辉,徐学宇,唐海,徐云根*
(1. 中国药科大学药学院,江苏 南京 211198;2. 天士力研究院化学药品开发中心,天津 300410)
翻译后修饰赋予了多肽、蛋白质和糖类等不同的结构和功能,是细胞生命活动不可或缺的调控机制,蛋白质可逆磷酸化是其中重要一部分。大多数细胞内蛋白质都会在一个或多个氨基酸位点上发生磷酸化修饰。蛋白质磷酸化程度的变化会改变蛋白质活性、稳定性、空间定位,以及蛋白质间的相互作用,使细胞对外界环境的变化作出精确的应答,进而调控细胞的增殖、分化、生长和凋亡等几乎所有的细胞进程。蛋白酪氨酸的磷酸化和去磷酸化这2个相反的过程分别由蛋白质酪氨酸激酶(PTK)和蛋白质酪氨酸磷酸酶(PTP)互相配合、精密调控,当这种平衡遭到破坏会诱发多种疾病,如炎症、肿瘤、自身免疫疾病等[1-2]。
含Src同源2结构域蛋白酪氨酸磷酸酶(SHP2)是由Ptpn11基因编码的非受体型蛋白酪氨酸磷酸酶,其在人体中广泛表达,特异性地催化细胞质中磷酸酪氨酸的去磷酸化过程。它是目前PTP家族中唯一被证实的原癌蛋白,在肿瘤细胞侵袭、转移、增殖、凋亡、耐药性等方面发挥重要调控作用,是一个理想的癌症干预靶标[3-4]。Ptpn11基因突变引起SHP2的超活化或表达上调与Noonan综合征、幼稚型单核粒细胞白血病、骨髓增生异常综合征及乳腺癌、肺癌、黑色素瘤等实体瘤密切相关[5-6]。针对SHP2开发其抑制剂成为肿瘤治疗的又一希望,在这一方向上已有大量研究,本文将对该靶点及其小分子变构抑制剂研究进展进行综述。
1 SHP2结构及参与的信号通路
SHP2磷酸酶的蛋白结构由3部分组成:2个串联的Src同源2结构域(SH2,包括N-SH2和C-SH2)、活性的催化位点结构域(即PTP),以及富含脯氨酸的无序C末端(见图1)。PTP酶活性严格依赖459位半胱氨酸。在基础状态下N-SH2与PTP结构域结合,阻断底物进入催化位点,使之处于封闭的自抑制构象,不能发挥去磷酸化功能[4,7]。SHP2的激活有以下2种方式。1)SH2与磷酸酪氨酸基序结合。可以是生长因子、细胞因子、炎症因子等刺激下SHP2的C端磷酸化的p-Tyr542和p-Tyr580,也可以是磷酸化的受体蛋白(如胰岛素受体底物-1(IRS-1))与SH2结构域特异性结合,使SHP2靠近底物,弱化N-SH2/PTP结构域之间的相互作用,打开自抑制构象[5,8]。2)PTP和N-SH2结构域的结合界面发生突变。这可能影响关键氨基酸残基的相互作用而不利于自抑制构象的形成和维持。例如,在白血病和一些实体瘤中发现的E76K突变,导致肥厚性心肌病的Q51E突变,还有PTP结构域中常见的Q506P突变等,都存在SHP2的过度激活[9-10]。
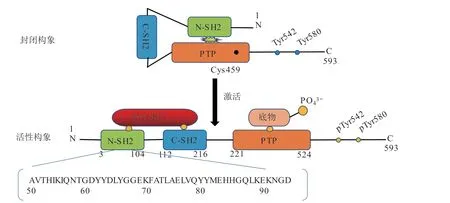
图 1 SHP2蛋白质序列及构象Figure 1 The protein sequence and conformation of SHP2
SHP2定位在细胞质中,传递来自多种受体酪氨酸激酶的细胞信号,调节了人体内多条信号通路,包括大鼠肉瘤蛋白(Ras)/细胞外调节蛋白激酶(ERK)通路、程序性死亡受体1(PD-1)/程序性死亡配体1(PD-L1)通路、磷脂酰肌醇3-激酶(PI3K)/蛋白激酶B(AKT)通路、Janus激酶(JAK)/信号传导及转录激活蛋白(STAT)通路、核因子κB(NF-κB)通路、肿瘤抑制蛋白-53(P53)通路等(见图2)。目前认为SHP2主要通过参与Ras/ERK和PD-1/PD-L1这2条通路来发挥作用[8,11]。在Ras/ERK通路中,SHP2是关键调控因子,位于Ras上游,几乎所有激活Ras的方式[比如细胞外刺激因子与受体酪氨酸激酶(RTK)、表皮生长因子受体(EGFR)结合,激活受体,进而介导的Ras激活]都需要经过SHP2来完成。一方面,SHP2被RTK激活,发挥其PTP活性,将细胞磷酸化信号衔接分子1(Gab1)去磷酸化,使Ras-GTP酶激活蛋白(Ras-GAP)激活并分离出Ras,增强其与效应蛋白丝氨酸/苏氨酸蛋白激酶(Raf)的联系;另一方面,激活的SHP2其C端酪氨酸发生磷酸化,募集生长因子受体结合蛋白2(Grb2)和sos基因编码的Ras激活蛋白(SOS),促进Ras激活,从而使整个ERK通路完全活化[6,12]。许多肿瘤疾病的发生都伴随Ras/ERK通路的过度活化,通过抑制SHP2阻断Ras激活,可以直接抑制某些肿瘤细胞的生长,治疗Ras驱动的人类癌症。在PD1/PD-L1通路中,SHP2是关键下游效应因子,被PD-1激活并募集到膜上,催化CD28(重组分化簇蛋白-28)共刺激受体、ZAP70(重组人Zeta链相关蛋白激酶-70)激酶去磷酸化,进而介导了Th1(辅助性T细胞-1)免疫中PD-1的抑制作用,在正常组织中是对自身的免疫保护,而在肿瘤中则促进了肿瘤细胞的免疫逃逸。抑制SHP2可以激活T细胞,增强免疫系统的功能,逆转在肿瘤微环境中的免疫抑制现象,从而清除癌细胞,故可作为PD-1、PD-L1抗体药物的辅助治疗[7,13]。总之,SHP2抑制剂同时具有化学治疗和免疫治疗2种功能,是一种具有双重活性的新型治疗药物。
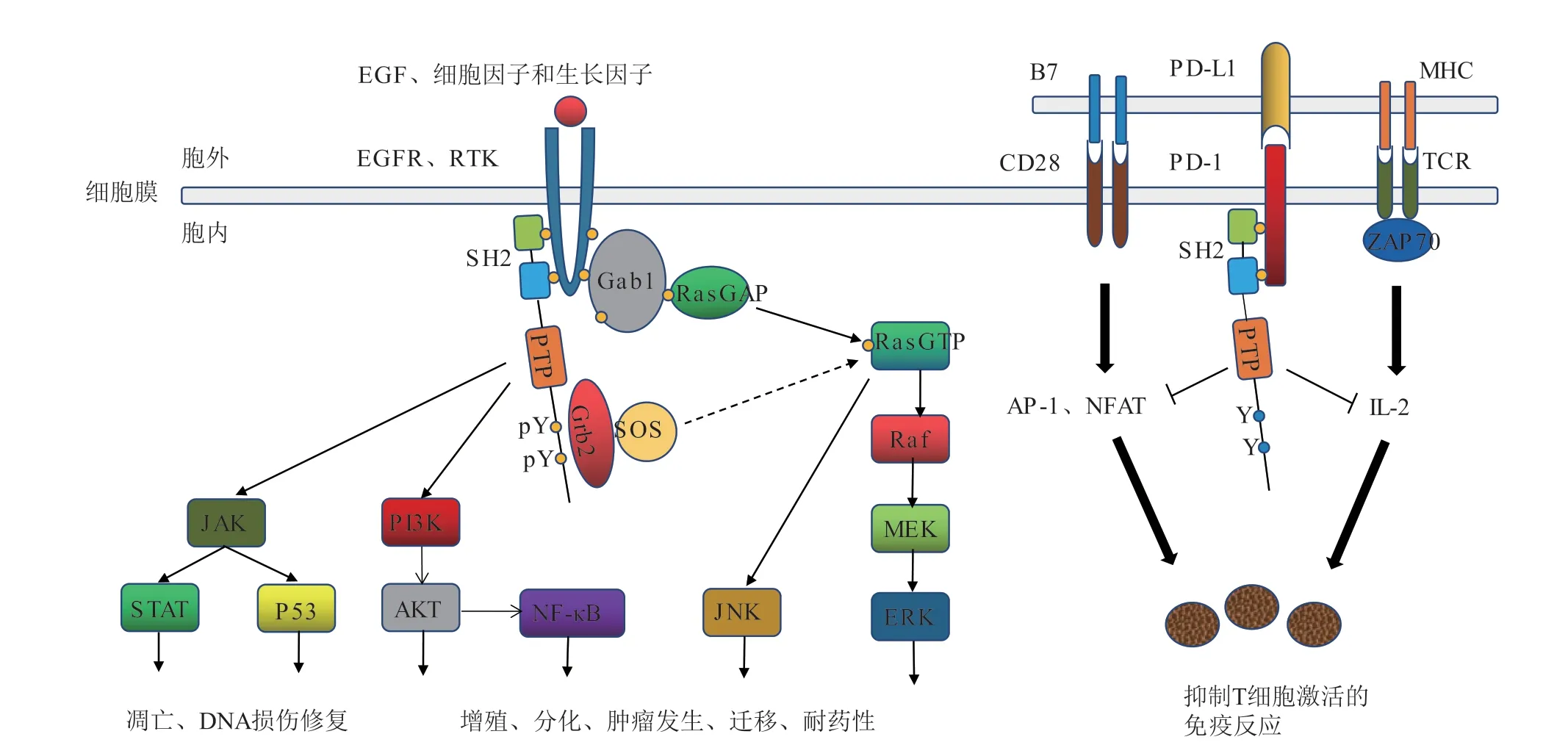
图 2 SHP2参与的信号通路及功能Figure 2 Signal pathways and functions with SHP2 involvement
2 SHP2小分子抑制剂
SHP2作为潜在的抗癌药物靶点引起了研究人员的广泛关注。传统的酶类抑制剂药物大多作用在催化位点,通过可逆或不可逆的方式与底物竞争结合在同一口袋来起到抑制作用。在早期,人们也曾开发过SHP2的催化位点抑制剂,例如含水杨酸、磺酸基团的一类化合物和某些天然产物[1-2]。但是在PTP家族中磷酸酶活性域高度保守,使催化位点抑制剂在高同源性的SHP1、SHP2、PTP1B中缺乏选择性,而且结构中大多含有模拟磷酸化底物与催化中心相互作用的极性和离子功能基团存在,导致催化位点抑制剂的细胞渗透性和生物利用度较差,加之本身酶抑制活性不高,使得这类药物难以开发成药[5,13]。University of Missouri的PHPS-1(1)、Aarden Pharmaceuticals Inc的I-A09(2),其 对SHP2的IC50分 别 为2.2和26.4 μmol · L-1。Cleveland Clinic Foundation的VQD-001(3)、VU University Medical Center的NSC-87877(4)对SHP1和SHP2的选择性差,还表现出了剂量依赖的细胞毒性、心脏和静脉毒性,导致催化位点抑制剂的研究被迫终止[2,7]。
后来,诺华团队在基于SHP2自抑制机制的磷酸酶测定研究中,使用近全长酶和PTP短片段进行高通量筛选以及识别不需要的催化位点,首次发现SHP2变构抑制剂并于2015年发表第1篇相关专利,2016年在《药物化学杂志》(JMC)上报道了SHP2变构抑制剂SHP099(5),其与活性中心以外的其他口袋相互作用来稳定或抑制酶构象的变化,使活性中心不能与底物结合而发挥功能。变构抑制剂不仅具有良好的SHP2抑制活性,且选择性高。SHP2的SH2结构域之间的连接子尤其是精氨酸残基的位置以及变构口袋大小均与SHP1不同(SHP2和SHP1的精氨酸残基位置分别为Arg111和Arg109,变构口袋大小分别为4.64×10-28m3和1.012×10-27m3),使得SHP2变构抑制剂对SHP1几乎没有抑制活性。此外,变构抑制剂成药性好、可口服利用,弥补了催化位点抑制剂的不足,开启了新的研究方向[3,11]。近年来的不断改进,使变构抑制剂的活性发生了质的飞跃,比如Revolution Medicines Inc的RMC-4550(6)和RMC-4630、MD Anderson Cancer Center的IACS-13909(7), 其对SHP2磷酸酶的IC50都在10 nmol · L-1以内[3,13]。针对该靶点目前还没有药物获批上市,进展最快的处于Ⅱ期临床阶段的药物,有诺华公司的TNO-155(8)、加科思的JAB-3068(9)和Revolution Medicines Inc的RMC-4630;处于Ⅰ期临床的有加 科 思 的JAB-3312、Relay Therapeutics的RLY-1971、MD Anderson Cancer Center的IACS-13909和Pfizer Inc的PF-07284892(见表1)。在研的15个变构抑制剂中,国内有5个,表现较为出色的以加科思的药物为代表,其在研发效率、临床进展、化合物活性等方面都走在了该领域的前沿。总之,SHP2变构抑制剂正在迅速起步,国内外竞争日渐激烈,该类药物将成为肿瘤治疗的又一重磅武器。
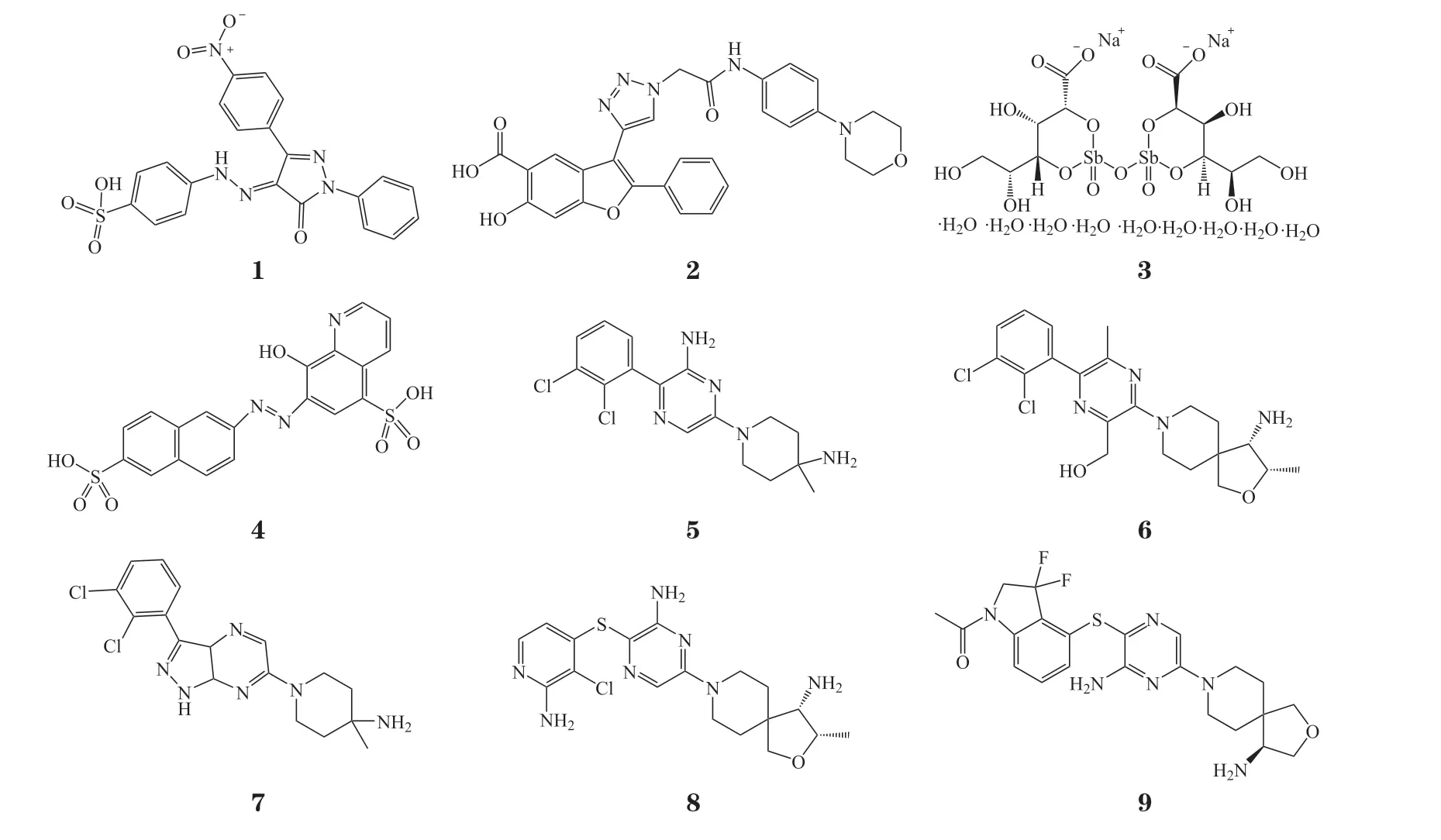

表 1 在研的SHP2抑制剂一览Table 1 List of SHP2 inhibitors under development
3 SHP2变构位点及构效关系分析
诺华团队使用Maestro中的SiteMap功能分析SHP2蛋白中可配位的口袋,揭示了3个潜在的小分子变构结合位点:1)位于N-SH2、C-SH2和PTP这3个结构域之间的“隧道”样变构位点,在所有变构位点中具有最大的内表面,是SHP099类化合物的结合口袋;2)离“隧道”约2×10-9m,位于N-SH2/PTP结构域界面的两侧的“门闩”变构位点,是SHP244类化合物的结合口袋;3)位于N-SH2/PTP结构域界面“门闩”另一面的“凹槽”结合位点,目前没有结合在该位点抑制剂报道[1,5]。
我们关注了SHP2变构抑制剂在研发历程中的结构变化,从构效关系的角度进行总结。这些抑制剂在结构上具有很高的相似度,基本都是诺华公司SHP099、TNO-155的结构类似物,都是结合在“隧道”样口袋的构象稳定剂。本文从二维结构的角度,将在研的SHP2小分子变构抑制剂的结构分为2类,即有2种通式:疏水区A、中心区B和极性区C(Ⅰ式),以及在A、B之间增加连接原子L(Ⅱ式)(见图3)。以下以诺华公司、Revolution Medicines Inc、加科思这3家具有代表性的公司为例进行详细介绍。
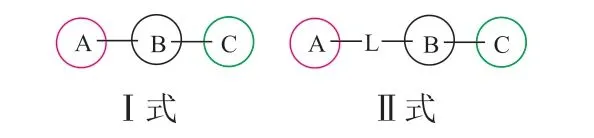
图 3 SHP2变构抑制剂结构通式Figure 3 The general structure formula of SHP2 allosteric inhibitors
从SHP099与SHP2的结合模式来看(见图4),主要有以下3部分作用力:1)极性区C的哌啶环上取代的氨基与Thr108、Phe113、Glu249残基的氢键相互作用以及与Glu249残基的离子键相互作用;2)中心区B的吡嗪环与Arg111的H-π相互作用,以及吡嗪环上取代的氨基与Glu250残基的氢键相互作用;3)疏水区A的二氯取代苯环与Pro491残基之间,以及哌啶环与His114残基之间存在的π-π堆积和疏水作用。这些作用力,使SHP099结合在N-SH2—C-SH2铰链区、C-SH2域和PTP活性域三者之间,稳定非活性构象。在“隧道”口袋两端分别为具有较大表面的、开放的溶剂可及区,为抑制剂的分子优化、衍生化提供了更多潜在的结合位点和空间结构变化的灵活性,或大或小的母核、不同类型的取代基都是允许的。
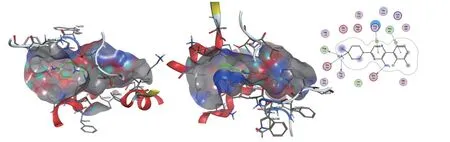
图 4 小分子SHP099与SHP2蛋白共晶结构Figure 4 The eutectic structure of small molecule SHP099 and SHP2 protein
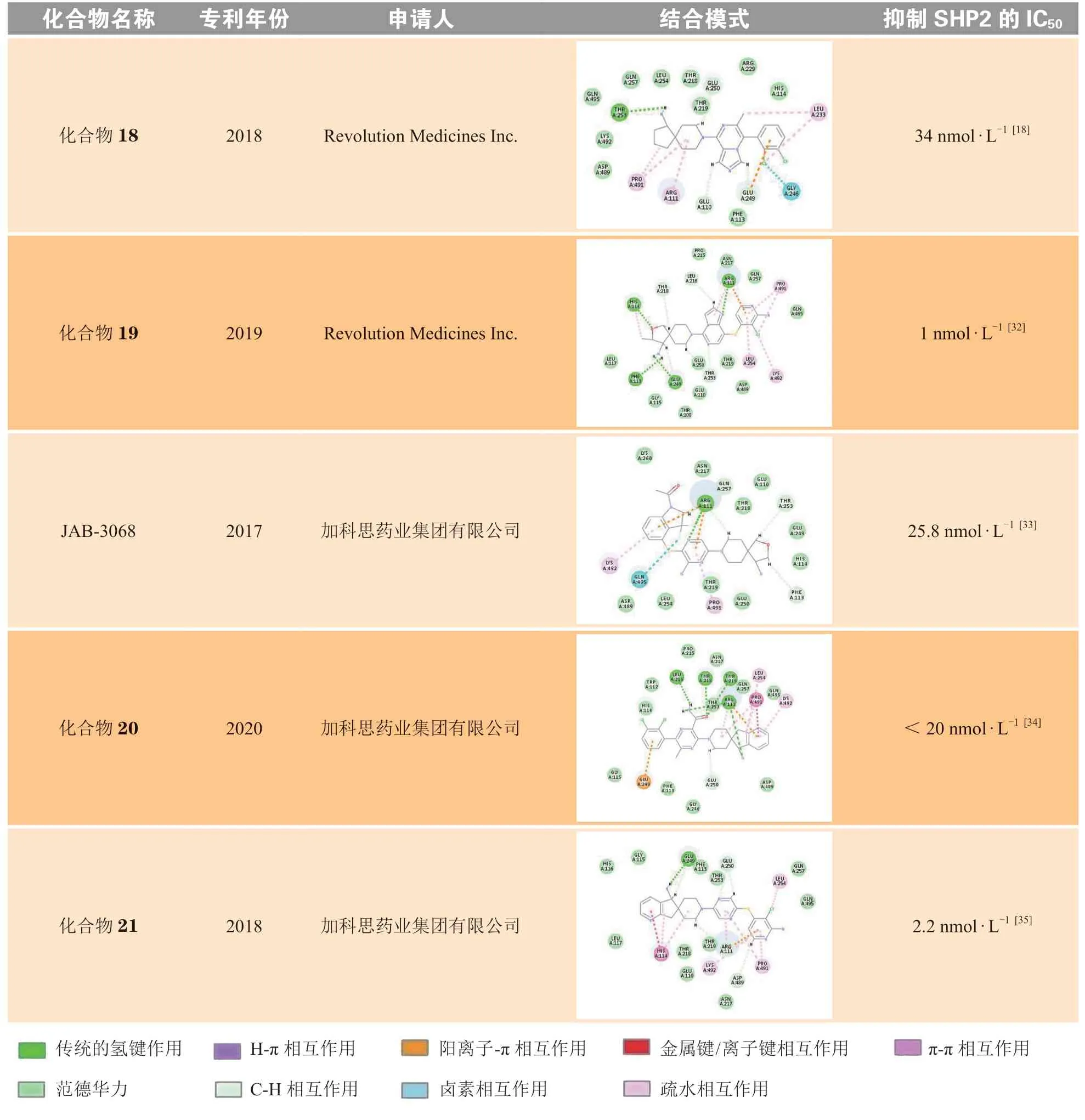
诺华在SHP099的结构基础上进行了一系列的结构修饰和改造,公开发表了共7篇化合物专利。笔者选取了每篇专利中活性最佳、结构具有代表性的化合物进行分子对接(见表2),将对接结果分别与SHP099进行对比分析。其中化合物10的极性区C为哌啶环,在哌啶环3位取代羟基可以与Glu110形成氢键作用,但对活性的提升不大。化合物11的疏水区A为二氯吡啶环,中心区B为吡嗪环,在A和B之间增加S原子,可增加化合物柔性,进而使化合物活性增加;将疏水区A的二氯取代苯替换为二氯取代吡啶,可以与Lys492形成π-π相互作用,但对活性的提升不大;极性区C哌啶环4位取代氨基替换为延长1个碳原子的氨甲基,对活性影响不大。TNO-155将极性区C的哌啶环衍生为螺[4·5]的螺环结构,螺环对取代的氨基具有一定的构象限制作用,增强了对氨基的空间定位,有利于形成氢键相互作用,进而提高化合物活性。化合物12将中心区B的氨基取代的吡嗪衍生为别嘌醇结构,别嘌醇烯醇互变的羰基虽然可以与Arg111形成氢键作用,但对活性的提升不大。化合物13与14的疏水区A部分中将吡啶环上2位取代的氯分别替换为甲氧基和氨基,其作为给电子基团,增强了吡啶环的电子云密度,进而增强了与Arg111、Pro491的H-π相互作用和阳离子-π相互作用,进而提高了化合物活性。诺华公司活性最好的分子为化合物15,其抑制SHP2酶活性的IC50为1 nmol · L-1,疏水区A为苯环,在苯环3位以酰胺键连接四氢吡啶并嘧啶酮结构,延长了疏水区的填充体积,增加了苯环外部的亲水性,通过与Thr253、Ser109形成氢键作用,以及与Glu249、Thr108形成离子键作用,显著提升了化合物活性。
Revolution Medicines参考了诺华SHP099系列化合物的结构骨架修饰和改造策略,公开发表了共5篇化合物专利。将每篇专利中代表性化合物的对接结果分别与SHP099进行对比,可以发现,RMC-4550将极性区C部分的螺环全碳骨架替换为2-氧杂-3-甲基取代的螺环,骨架中的氧原子可以与His114形成氢键作用,使化合物活性略有提升。将SHP099中心区B的吡嗪环的2位取代氨基替换为对位甲基和羟甲基,可以与Thr218残基形成氢键作用以及与Thr219形成离子键相互作用,使化合物活性明显提升。对比RMC-4550,化合物16将中心区B取代的甲基替换为羟基,化合物17将取代的甲基去除,减弱了疏水区A与变构口袋的疏水相互作用,进而使活性略微降低。化合物18的中心区为环合的咪唑并吡嗪结构,可以与Arg111形成氢键作用,但对活性的提升不大。Revolution Medicines活性最好的化合物为化合物19,其抑制SHP2酶活性的IC50为1 nmol · L-1。该化合物将中心区B换成了含更多氮原子的咪唑并嘧啶的双环结构,增加了与B部分Arg111的氢键作用,同时极性区C也与His114形成氢键作用力,从而获得较高的活性和选择性。
加科思在诺华SHP099系列化合物的结构基础上进行了修饰和改造,公开发表了共3篇化合物专利。将每篇专利中的代表性化合物分别与SHP099的结合模式进行对比分析,结果显示,JAB-3068将疏水区A的二氯取代苯衍生为二氟和甲酰基取代的苯并吡咯的双环结构,氟原子与Gln495形成卤素相互作用,疏水区A协同中心区B的氨基吡嗪结构,增强了化合物与Arg111形成的氢键作用和阳离子-π相互作用,使活性略有提升。对比SHP099,化合物20将中心区B的2位氨基取代吡嗪替换为对位甲基和氨甲酰基取代的吡嗪,可同时与Leu216、Thr218、Thr219形成氢键作用,提高了化合物活性。加科思公司活性最好的化合物为化合物21,其抑制SHP2酶活性的IC50为2.2 nmol · L-1。该化合物将极性区C从SHP099的氨基取代的哌啶环,优化为TNO-155的二元螺环,进一步扩增为环戊烷并吡啶的三元螺环,主要通过增加C部分的分子柔性和疏水体积,增强了定位效应,并引入具有芳香性的吡啶结构,可与His114形成π-π相互作用,从而获得较高活性。
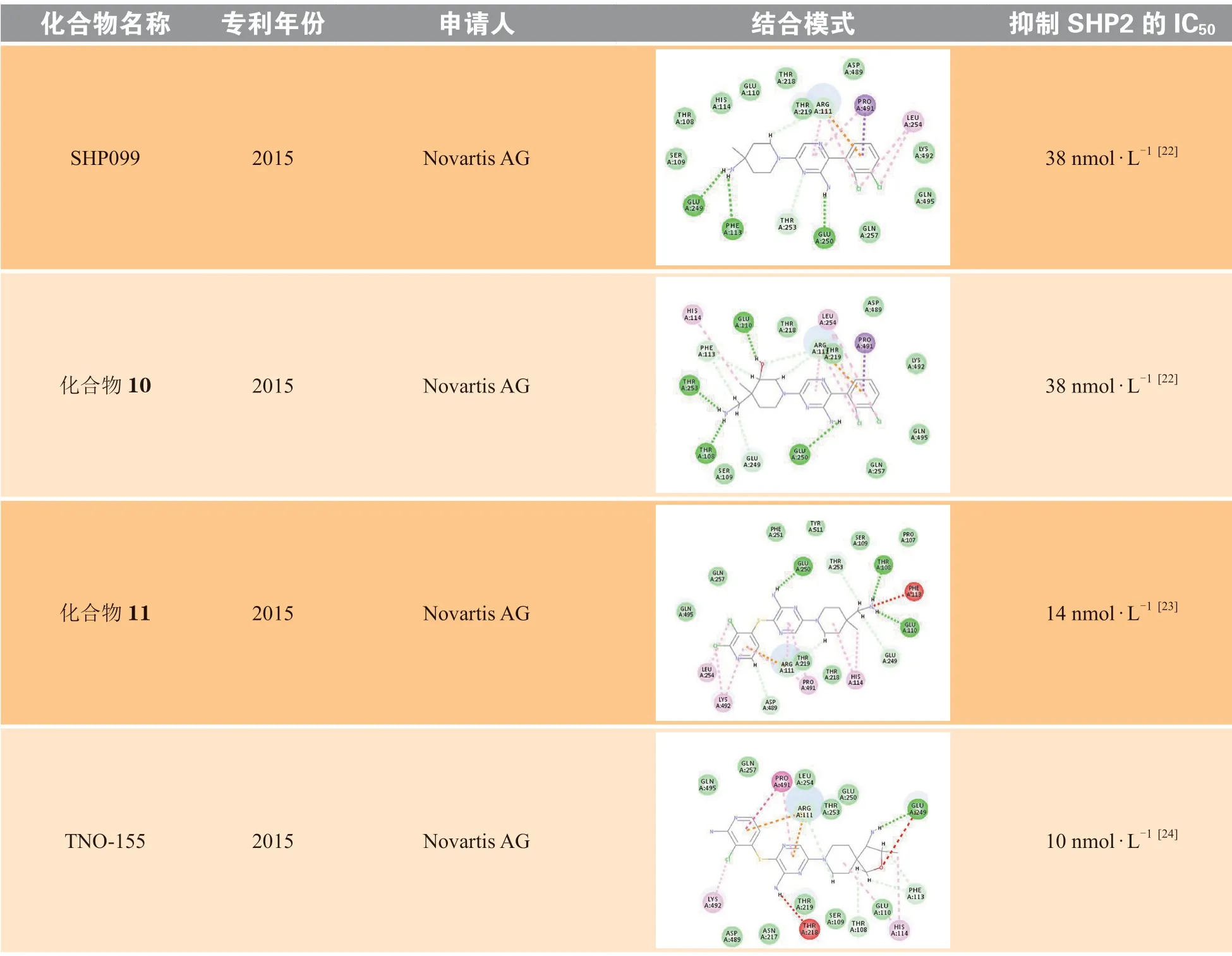
表 2 专利中的代表化合物及其SHP2抑制活性Table 2 The representative compounds and their inhibitory activities against SHP2 in patents

综上所述,疏水区A和极性区C是影响化合物活性的关键部分。与疏水区A相互作用的口袋外部有亲水性结合位点,符合内部疏水、外部亲水的规律,为疏水部分的优化提供了指导。与极性区C相互作用的是一个空间较大的两性结合区,C部分在整体上是一个起到定位作用的疏水骨架,其中包含能与蛋白口袋形成氢键相互作用的亲水性基团,这是维持化合物活性所必需的。对疏水区A和极性区C这两部分进行结构改造,可以高效获得活性各异的候选药物分子。虽然中心区B部分结构变化对活性影响不大,但中心区的吡嗪环从没有氨基取代到有氨基取代,化合物对SHP2的IC50由5.7 μmol · L-1变为70 nmol · L-1,因此B环的氢键作用也是确定且必不可少的。此外,中心区B是与Arg111相互作用的主要结构,因此B部分的变化还可能与化合物选择性有关。
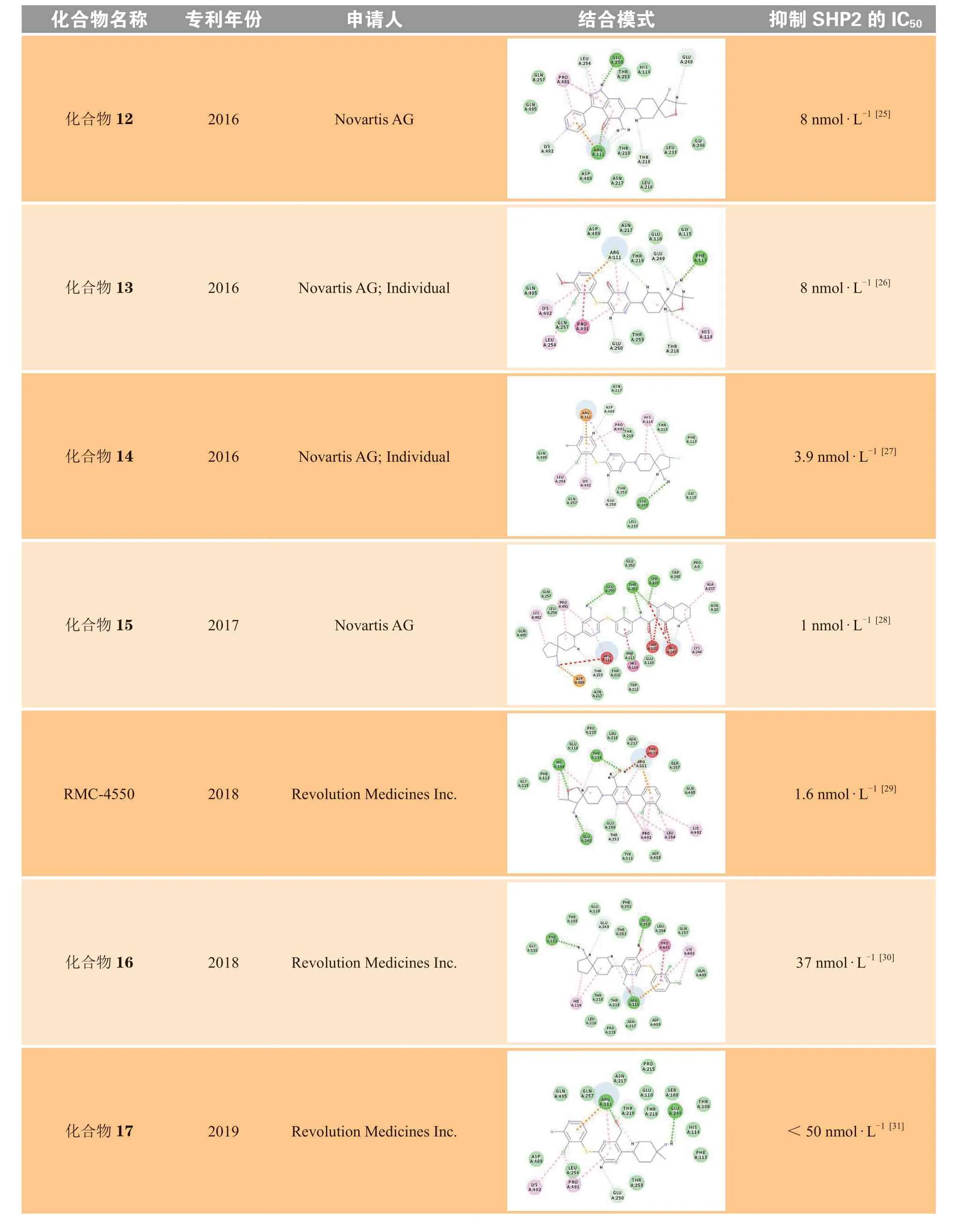
续表2
续表2
4 结语与展望
SHP2是一个具有潜力的癌症治疗靶点,从它的调控机制和药物临床数据来看足以起到治疗效果,同时SHP2也被视为耐药性发展的共同节点,SHP2抑制剂在联合用药和替代药物的使用方面有很大的优势和空间。然而,SHP2通路及其抑制剂也存在一些问题和争议,例如Revolution Medicines Inc开展的临床试验表明RMC-4630对RAS突变(KRASmt、KRASG12C)的非小细胞肺癌(NSCLC)患者具有一定疗效,疾病控制率分别达到67%和75%,而Chen等[11]研究表明SHP2抑制剂对具有组成型激活RAS信号的人乳腺癌细胞SUM52(KRASG12V)、MDA-MB-231(KRASG13D)或黑色素瘤细胞A2058(BRAFV600E)突变株的生长和增殖没有影响。SHP2抑制剂通过构象选择机制仅与闭合状态的SHP2相结合,致癌性SHP2突变可能影响结合口袋的形态或与小分子的作用力,将极大地降低抑制剂的亲和力和活性,如SHP099与E76K、T253M、Q257L突变型的亲和力相比于野生型降低至0.1% ~ 1.0%,抑制活性与突变激活的磷酸酶活性成反比[36]。因此,SHP2抑制剂对不同的突变细胞和不同的突变类型的有效性是不同且有限的。靶向不同变构位点,开发具有针对性、更强效的药物或尝试联合用药的方案可能是克服癌症突变的一种手段[1,9]。另一方面,SHP2在细胞通路中的位置靠近上游,影响范围是广泛的,但在各个通路中的确切机制仍不是很清楚。例如,SHP2可正向调节血小板衍生因子(PDGF)和胰岛素样生长因子(IGF)诱发的AKT激活,但在EGFR激活的PI3K/AKT通路中,SHP2具有不同的调控作用,在胶质瘤细胞中诱导AKT激活,而在成纤维细胞中诱导AKT抑制。在JAK/STAT通路中,SHP2也具有双重作用[4,10]。前文提到,抑制SHP2活性对多种肿瘤具有良好的疗效,然而从另外一些角度评价这类抑制剂,又存在一些问题。例如在肝癌和软骨瘤中激活SHP2表现出肿瘤抑制功能,以及在CD4+T细胞中抑制SHP2活性能促进黑色素瘤的肺转移,因此抑制SHP2是否存在可引起其他肿瘤疾病的风险是未知的[6-7]。SHP2的作用具有细胞类型和组织的特异性,需要开展更深入的机制研究,以阐明其在不同细胞与肿瘤微环境中的确切功能,从而更好地发挥SHP2变构抑制剂的治疗效果、降低其毒副作用,从而使这类药物更加安全有效地应用于肿瘤患者。
猜你喜欢
中风与神经疾病杂志(2022年9期)2022-10-19
湖北农业科学(2022年11期)2022-07-18
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
分析化学(2017年12期)2017-12-25
中文信息(2017年2期)2017-04-13
江苏农业科学(2016年11期)2017-03-21
智能计算机与应用(2016年4期)2016-09-26
中学化学(2015年12期)2016-01-19
中学化学(2015年12期)2016-01-19
