
新型大位阻氮杂环卡宾咪唑啉盐的合成
2022-04-24 09:53陆辉扬曹育才
上海塑料 2022年2期
沈 安,陆辉扬,曹育才
(1.上海化工研究院有限公司,上海 200062;2.聚烯烃催化技术与高性能材料国家重点实验室,上海 200062;3.上海市聚烯烃催化技术重点实验室,上海 200062)
0 前言
咪唑(啉)型氮杂环卡宾的应用广泛,不仅催化活性高,而且对空气、水的稳定性也好。通过氮原子上引入取代基可以进一步强化咪唑(啉)型氮杂环卡宾的电子特性与空间性能,在不同类型的催化反应中取得较好效果。利用不同结构苯基取代的咪唑(啉)型氮杂环卡宾作为配体能高效催化碳碳偶联反应[12-15];针对特殊的反应情况,可选用更大空间位阻的配体[16],并通过远端电子效应调节机制,进一步提升催化效果[17]。ORGAN M G等[18]针对氮原子上苯环的取代基团进行改造,拓展了配体的种类;而针对氮杂环上C4和C5位取代基团的改造,则有利于Negishi反应中的β-H消除[19]。
此外,针对氮杂环卡宾的环骨架还可以在N1和N3位引入非对称取代基团或构型可变的大位阻基团,使氮杂环卡宾配体能更好地适应不同催化循环阶段对位阻的不同需求[20-23]。
目前,咪唑(啉)型氮杂环卡宾结构上的修饰集中于氮原子所连苯环的取代基团上,且均为烷基或烷芳基团,种类还不够丰富。笔者从咪唑(啉)型氮杂环卡宾结构设计出发,合成了大位阻咪唑(啉)型氮杂环卡宾,进一步丰富该类配体的结构种类。在取代基团的选择上,引入含有氮、氧等杂原子基团,同时充分考虑取代基的位阻效应,优先选择了含杂原子的五元环吡咯烷取代基和六元环吗啉取代基。
1 实验部分
1.1 主要试剂
2,6-二氯硝基苯(纯度96%)、钯碳(Pd/C,质量分数5%)、硼烷二甲硫醚-四氢呋喃(DMSB,浓度2 mol/L)、原甲酸三乙酯(纯度99%)、吡咯烷(纯度99%),安耐吉化学试剂有限公司;
吗啉(纯度98%)、丁二酮(纯度98%)、氯甲基乙醚(纯度95%)、三甲基氯硅烷(TMSCl,纯度98%)、三乙胺(纯度99%),上海九鼎化学科技有限公司;
草酰氯(纯度98%),国药集团化学试剂有限公司;
吡啶(Py,纯度99%)、4-二甲胺基吡啶(DMAP,纯度99%),梯希爱(上海)化成工业发展有限公司;
乙二醛(纯度98%)、无水甲酸(纯度99%)、硼氢化钠、四氢铝锂、无水硫酸钠、氯化钠、氯化钾(分析纯),江苏强盛功能化学股份有限公司;
二氯甲烷、甲苯(TOL)、乙酸乙酯(EA)、四氢呋喃(THF)、正己烷(HEX)、甲醇、乙醇(分析纯),上海迈瑞尔化学技术有限公司。
1.2 主要设备及仪器
核磁共振谱仪,JNM-ECZ500,频率为500 MHz,日本电子株式会社;
集热式恒温加热磁力搅拌器,HF101S 型,巩义市予华仪器有限公司;
循环水式真空泵,SHB-III型,上海卫凯仪器设备有限公司;
旋转蒸发仪,N-1210B 型,东京理化器械株式会社。
1.3 实验方法
设计的氮杂环卡宾咪唑盐合成路线见图1。

图1 氮杂环卡宾咪唑盐合成路线
1.3.1 2,6-二吗啉硝基苯7a的合成
在200 mL的圆底烧瓶中加入2,6-二氯硝基苯(6.99 g,36.4 mmol)和吗啉(90.60 g,1 040 mmol),混合后置于120 ℃油浴反应7 h。反应结束后,将反应液倒入50 mL CH2Cl2中,用水洗涤,用10 mL CH2Cl2萃取水相3次,合并有机相后用无水硫酸钠干燥,旋蒸除去溶剂,然后加入50 mL正己烷加热回流并滤去不溶物,滤液用正己烷重结晶得到9.07 g橙色固体,产率为85%。核磁共振氢谱(1H-NMR)(500 MHz,CDCl3)化学位移(δ):7.39 (三重峰(t),偶合常数(J)=8.1 Hz,1H),7.02 (双重峰(d),J=8.2 Hz,2H),3.81~3.69 (多重峰(m),8H),3.03~2.88 (m,8H)。
1.3.2 2,6-二吗啉苯胺8a的合成
在1 L的圆底烧瓶中加入2,6-二吗啉硝基苯(5.10 g,17.4 mmol)、乙醇(350 mL)、5%(质量分数,下同)Pd/C(1.85 g,0.87 mmol),然后用N2置换3次,再用H2(0.1 MPa)置换3次,最后在H2(0.2 MPa)的氛围中保持30 ℃并剧烈搅拌8 h。反应液经硅藻土过滤除去Pd/C,滤液旋蒸至40 mL,然后置于-30 ℃的环境下缓慢加入80 mL蒸馏水,析出固体,过滤干燥后得到3.44 g白色固体,产率为75%。1H-NMR(500 MHz,CDCl3)δ:6.88~6.66 (m,3H),4.26 (宽峰(bs),2H,NH),4.07~3.56 (m,8H),3.09~2.81 (m,8H)。
1.3.3N,N′-双(2,6-二吗啉苯基)丁二酮亚胺9a的合成
在25 mL的三口烧瓶中加入化合物8a(1.05 g,4 mmol),用N2置换3次,再依次加入丁二酮(2 mmol)、10 mL甲醇和催化量的甲酸,然后室温搅拌72 h。反应结束后抽滤,并用10 mL甲醇洗涤滤饼,得0.7 g黄色固体9a,产率为60%。1H-NMR (500 MHz,CDCl3)δ:7.05 (t,J=8.1 Hz,2H),6.73 (d,J=8.2 Hz,4H),3.75 (dddd,J=35.0 Hz、11.0 Hz、6.3 Hz、2.8 Hz,16H),3.31 (ddd,J=12.0 Hz、6.5 Hz、3.0 Hz,8H),2.69 (ddd,J=11.8 Hz、6.4 Hz、2.7 Hz,8H),1.92 (单峰(s),6H)。核磁共振碳谱(13C-NMR)(125 MHz,CDCl3)δ:166.80,142.01,138.68,124.51,112.67,67.46,50.70,15.01。
1.3.4N,N′-双(2,6-二吗啉苯基)-2,3-丁二胺10a的合成
在25 mL的三口烧瓶中加入9a(0.23 g,0.4 mmol),用N2置换3次,再依次加入3 mL THF、1 mL TOL和1 mL DMSB溶液(2 mmol),然后加热回流反应7 h,溶液变为澄清透明。反应液冷却至0 ℃,边搅拌边缓慢滴加甲醇进行淬灭反应至无气泡产生为止,再用20 mL二氯甲烷萃取3次,有机相用无水硫酸钠干燥,旋蒸得0.21 g固体10a,产率为88%。1H-NMR (500 MHz,CDCl3)δ:6.91~6.64 (m,6H),5.22 (bs,2H,NH),3.75 (t,J=4.6 Hz,16H),3.53 (s,4H),2.91 (t,J=4.6 Hz,16H),1.24 (s,6H)。13C-NMR (125 MHz,CDCl3)δ:142.64,137.55,120.12,115.80,67.59,51.48,44.92,29.80。
1.3.5N,N′-双(2,6-二吗啉苯基)-2,3-二甲基咪唑啉鎓氯化物11a的合成
在10 mL双口圆底烧瓶中依次加入化合物10a(2.44 g,4.2 mmol),NH4Cl(0.2 g,4.5 mmol),用N2置换3次后再加入原甲酸三乙酯(1 g,7 mmol),然后升温至110 ℃反应2 h。反应液用二氯甲烷溶解后过滤除去不溶物,滤液自然结晶,过滤并用1 mL乙醚洗涤,滤饼干燥至恒重得到1.79 g白色固体11a,产率为68%。1H-NMR (500 MHz,CDCl3)δ:8.19 (s,1H),7.52 (t,J=8.0 Hz,2H),7.33 (d,J=8.0 Hz,2H),7.23 (d,J=8.0 Hz,2H),5.51 (四重峰(q),J=5.5 Hz,2H),3.94 (dtq,J=14.5 Hz、9.0 Hz、3.0 Hz,8H),3.59 (s,8H),3.27 (ddd,J=11.5 Hz、6.0 Hz、3.0 Hz,4H),2.96~2.84 (m,12H),1.37 (d,J=6.0 Hz,6H)。13C-NMR (125 MHz,CDCl3)δ:157.63,149.32,148.41,131.39,125.25,121.40,120.08,67.09,66.92,63.90,55.00,52.47,19.58。
1.3.6 2,6-二吡咯烷硝基苯7b的合成
在200 mL圆底烧瓶中加入2,6-二氯硝基苯(3.84 g,20.0 mmol)和吡咯烷(51 mL,600 mmol)混合物置于120 ℃油浴反应7 h。反应结束后,将反应液倒入50 mL CH2Cl2中,用水洗涤,用10 mL CH2Cl2萃取水相3次,合并有机相后用无水硫酸钠干燥,旋蒸除去溶剂,然后加入50 mL正己烷加热回流并滤去不溶物,滤液用正己烷重结晶得到5.05 g固体7b,产率为96%。1H-NMR (500 MHz,CDCl3)δ:7.08 (t,J=8.3 Hz,1H),6.33 (d,J=8.3 Hz,2H),3.30~3.13 (m,8H),1.96~1.80 (m,8H)。
1.3.7 2,6-二吡咯烷苯胺8b的合成
在1 L圆底烧瓶中加入2,6-二吡咯烷硝基苯(4.55 g,17.4 mmol)、乙醇350 mL、5%Pd/C(1.85 g,0.87 mmol)。将烧瓶先用N2置换3次,再用H2(0.1 MPa)置换3次,最后置于H2(0.2 MPa)氛围中并保持30 ℃剧烈搅拌8 h。过滤所得悬浮液,滤液除去溶剂得到浅黄色固体3.67 g,将其置于500 mL锥形瓶中并加入20 mL乙醇,通过加热使其全部溶解,然后再置于-30 ℃的环境下并缓慢加入80 mL蒸馏水,析出大量固体,过滤干燥后得到3.54 g白色固体8b,产率为88%。1H-NMR (500 MHz,CDCl3)δ:6.83~6.59 (m,3H),4.02 (bs,2H,NH),3.07 (td,J=6.4 Hz、5.2 Hz、2.6 Hz,8H),1.98~1.82 (m,8H)。
1.3.8N,N′-双(2,6-二吡咯烷苯基)乙二酰胺9b的合成
在N2氛围下向25 mL三口瓶中依次加入化合物8b(0.5 g,2.16 mmol)、少许分子筛、Py(0.2 mL,1 mmoL)、催化量DMAP和THF (7 mL),然后降温至-5 ℃并缓慢滴加草酰氯(0.13 g,1 mmol),再逐渐恢复至室温反应9 h。向反应液中滴加几滴甲醇淬灭草酰氯,然后滤去不溶物,滤液用10 mL二氯甲烷稀释并用水洗涤3次,收集有机相后用无水硫酸钠干燥,旋蒸除去溶剂后用柱层析纯化(V(HEX)∶V(EA)=1∶2)得到0.25 g黄色固体9b,产率为48%。1H-NMR (500 MHz,CDCl3)δ:8.91 (bs,2H,NH),7.07 (t,J=8.2 Hz,2H),6.44 (d,J=8.2 Hz,4H),3.32~2.91 (m,16H),1.97~1.75 (m,16H)。13C-NMR (125 MHz,CDCl3)δ:157.66,148.77,128.54,124.53,116.24,67.47,52.03。
1.3.9N,N′-双(2,6-二吡咯烷苯基)乙二胺盐酸盐10b的合成
在N2氛围下称取9b(0.2 g,0.38 mmol)加入25 mL反应瓶中,再加入DMSB溶液(0.14 g,1.9 mmol)和THF (5 mL),然后在回流温度下反应8 h。反应液冷却至0 ℃,边搅拌边缓慢滴加1 mol/L HCl 至pH<4,然后用质量分数为10%的NaOH溶液调节至pH>8,再用50 mL二氯甲烷稀释后水洗3次,有机相用无水硫酸钠干燥,旋除45 mL溶剂,再边剧烈振荡边滴加几滴HCl至出现大量白色固体,过滤后得85.4 mg产物10b,产率为40%。1H-NMR (500 MHz,CDCl3)δ:6.92~6.50 (m,6H),4.93 (bs,2H,NH),3.17 (m,4H),3.03 (dq,J=9.7 Hz、6.1 Hz、4.8 Hz,16H),2.04~1.72 (m,16H)。13C-NMR (125 MHz,CDCl3)δ:141.64,137.03,119.87,112.71,50.98,44.93,24.39。
1.3.10N,N′-双(2,6-二吡咯烷苯基)咪唑啉鎓氯化物11b的合成
在10 mL双口圆底烧瓶中依次加入化合物10b (1.5 g,4.2 mmol)和NH4Cl (0.2 g,4.5 mmol),用N2置换3次后再注入原甲酸三乙酯(1 g,7 mmol),然后升温至110 ℃反应2 h。将反应液用二氯甲烷溶解后过滤除去不溶物,滤液自然结晶,过滤并用1 mL乙醚洗涤,干燥至恒重得到1.23 g粉色固体11b,产率为68%。1H-NMR (500 MHz,CDCl3)δ:7.77 (s,1H),7.28~7.25 (m,2H),6.81 (d,J=8.2 Hz,4H),4.64 (s,4H),3.16 (t,J=6.3 Hz,16H),1.95 (t,J=6.4 Hz,16H)。13C-NMR (125 MHz,CDCl3)δ:160.41,147.97,130.57,130.48,122.46,113.88,113.84,53.53,51.74,25.48。
2 结果与讨论
从咪唑(啉)型氮杂环卡宾结构设计出发,引入具有电子效应调节能力的大位阻基团吗啉和吡咯烷,分别通过5步反应得到最终产物11a和11b,其1H-NMR见图2、图3。由于反应路线涉及大位阻基团,且引入的吗啉和吡咯烷基团电负性相对更大,因此整体反应难度较大、副产物多、产率不高。通过改变合成路线,优化合成方法,最终分别以23%和11%的总产率得到新型大位阻取代氮杂环卡宾咪唑啉盐。
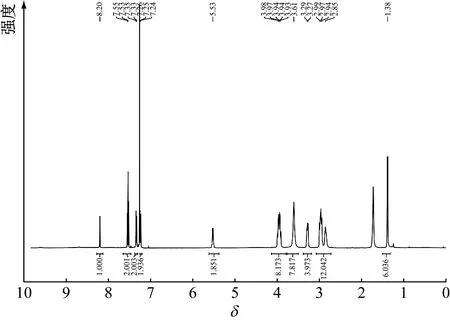
图2 化合物11a的1H-NMR

图3 化合物11b的1H-NMR
2.1 化合物11a的合成路线优化
按照最初的合成路线设计,化合物9a可参考文献方法直接成环得到氮杂环卡宾咪唑盐,但是,由于9a在有机溶剂中的溶解性极差,导致其在现有的反应条件下均不发生反应(见表1)。针对该情况修改了合成路线,先将9a还原成二胺10a,然后再环化成氮杂环卡宾咪唑啉盐。对还原反应条件进行优化,结果见表2。

表1 化合物9a环化反应体系筛选

表2 化合物9a还原反应体系筛选
由表2可以看出:选用NaBH4作为还原试剂时不发生反应;选用LiAlH4时,还原性过强,产物以8a为主;采用DMSB/THF体系可以顺利发生反应,进一步优化反应溶剂,最终以88%产率得到目标产物。
得到化合物10a后可以顺利进行环化反应得到相应氮杂环卡宾咪唑啉盐,反应总产率为23%。
2.2 化合物11b的合成路线优化
8b的稳定性差,特别是在醇类溶剂中极易变坏,反应要特别注意保护。8b和二醛的缩合反应较难发生,通过尝试多种反应条件的优化策略,即优化溶剂种类——乙醇、正丙醇、TOL,优化催化酸——甲酸、乙酸,优化反应时间——24 h、48 h、72 h,溶剂除水——加分子筛,优化反应温度——25 ℃、40 ℃、60 ℃、回流温度,均无法得到目标二亚胺产物。考虑到引入的吡咯烷基团可能增强苯胺化合物的碱性,影响正常的酸催化脱水缩合反应过程,从而可能导致乙二醛和8b只发生分子内的类Cannizzaro反应(见图4)。
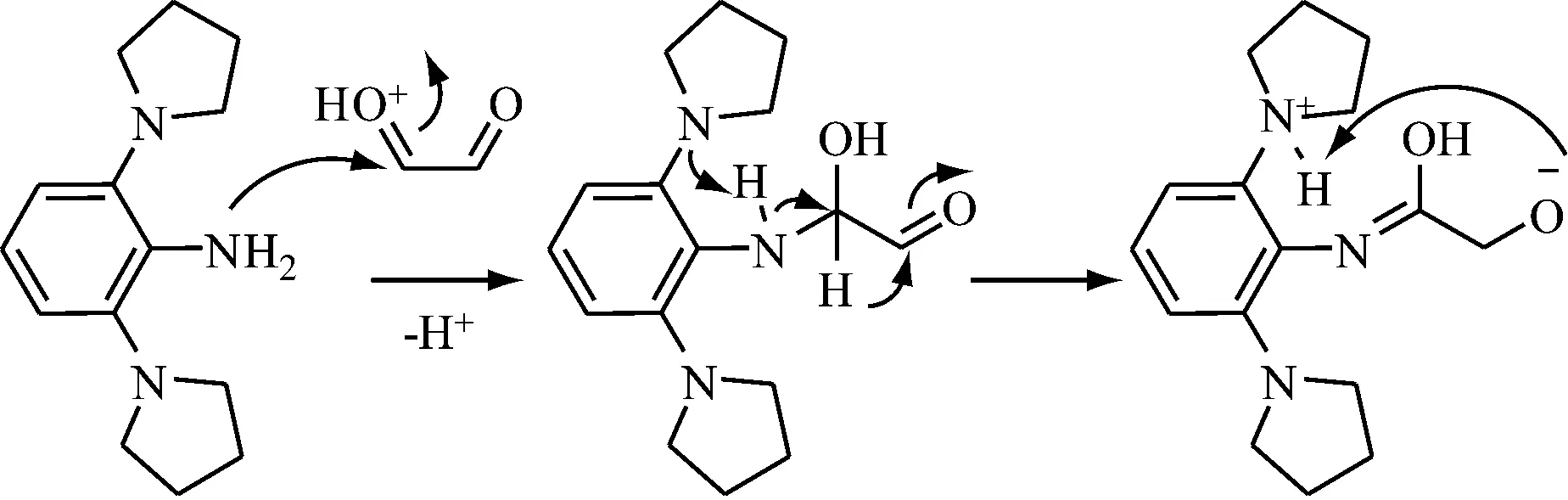
图4 推测化合物8b与乙二醛可能的反应机理
因此,对含有吡咯烷基的化合物尝试更换合成路线,考虑使用草酰氯和8b先制备酰胺,然后再通过酰胺还原的方法得到二胺。对制备酰胺的反应条件进行优化(见表3),选用二氯甲烷/三乙胺体系,反应复杂无主产物;选用THF/三乙胺体系,反应以单酰化产物为主,目标产物的产率很低;选用THF/Py体系,反应进行较为彻底,虽然有单酰化产物,但分离产率可达35%;若使用DMAP作为催化剂,可加快反应速率,进一步提升反应产率至48%。
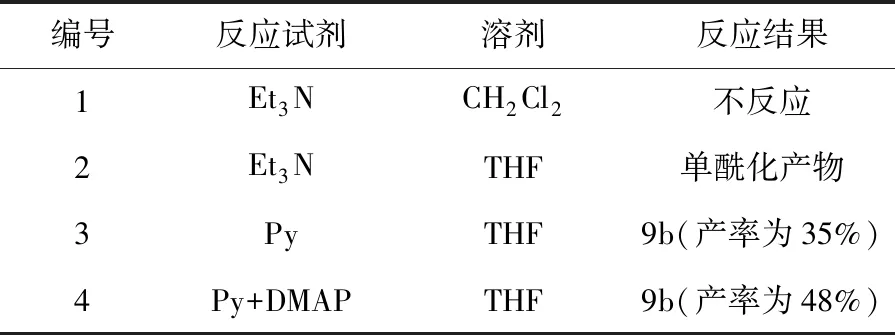
表3 化合物8b酰化反应体系筛选
2.3 氮杂环卡宾咪唑(啉)盐的应用
氮杂环卡宾咪唑(啉)盐不仅可以作为小分子催化剂直接催化反应[24],也可以作为配体与过渡金属配位后参与催化反应[25]。
PATRA A等[26]利用氮杂环卡宾咪唑(啉)盐实现芳香醛或脂肪醛与乙烯基磷酸酯分子间的Stetter反应。LIU J L等[27]报道了氮杂环卡宾咪唑(啉)盐催化2-硝基半乳糖糖苷化反应,可为糖肽的合成提供新方法。该类反应需要大位阻催化剂作为离去基团,而笔者合成的氮杂环卡宾咪唑(啉)盐具有更大的位阻。
在过渡金属催化领域,YENISARI B等[28]合成了二茂铁取代的氮杂环卡宾咪唑(啉)盐,可与金属钯形成稳定配合物。DENISOV M S等[29]则报道了金刚烷取代的氮杂环卡宾咪唑(啉)氯化钯盐,可实现碳氢键活化构建碳碳键。笔者合成的氮杂环卡宾咪唑(啉)盐具有类似结构,也有望与多种过渡金属配位形成配合物,用于催化反应,后续将继续深入研究。
3 结语
以新结构氮杂环卡宾的合成为目标,在氮原子所连苯环上引入大位阻吗啉和吡咯烷基团,成功实现2种新型氮杂环卡宾咪唑啉盐的合成,通过反应条件的筛选与优化,总产率分别为23%和11%。作为新结构的氮杂环卡宾配体,该类咪唑啉盐不仅具有较大的空间位阻,而且引入的吗啉和吡咯烷取代基有可能在催化反应中调节电子效应,为催化反应的配体选型提供了更丰富的可能性。
猜你喜欢
大连民族大学学报(2021年5期)2021-11-15
科学技术与工程(2020年34期)2021-01-08
世界农药(2020年12期)2021-01-04
世界农药(2019年4期)2019-12-30
农药科学与管理(2019年8期)2019-11-23
新课程·下旬(2019年7期)2019-09-17
肉类研究(2019年8期)2019-09-10
农药科学与管理(2019年12期)2019-05-20
农产品加工(2019年4期)2019-01-06
发明与创新·中学生(2017年11期)2017-12-07
