
美国非处方药专论制度改革分析及对我国的启示*
——基于社会性规制均衡视角
2022-04-27 06:45付瑞枫茅宁莹董晨东
中国卫生事业管理 2022年4期
付瑞枫,茅宁莹△,董晨东
(1.中国药科大学商学院,江苏 南京 211198;2.华润三九医药股份有限公司 )
社会性规制是政府为控制负外部性和可能会影响人身安全健康的风险,而采取的行动,如制定标准、禁止及限制特定行为等。自20世纪初社会性规制兴起之时开始,与人民健康息息相关的食品和医药行业就是重点规制领域[1]。我国也在药品的研发、生产、流通、使用、上市后不良反应监测等各个环节制定了一系列严格的法律法规体系和技术支撑体系,以保证我国药品质量安全水平,保护公众健康。
由于药品监管规制同时注重“保护人民健康”与“促进产业创新”,我国在推进药品监管规制过程中常常陷入是放还是松的两难困境。因此,政府在药品领域进行社会性规制时,要考虑如何把简政放权与合理规制结合起来,实现创新性与安全性的均衡[2]。
我国非处方药(Over-the-Counter, OTC)的监管中也存在同样的规制均衡考量。由于非处方药涉及人民群众范围广且销售门槛低,2020年以前,我国药品监管部门在非处方药的审评上设置了较为严格的审评程序,通常非处方药在申报后3~5年才可能获批上市[3]。此举虽可保证非处方药的安全性,却在一定程度上削弱了企业创新积极性,制约了我国非处方药行业的进一步发展,因此我国也在持续探索如何能够在保障安全性的前提下,简化非处方药注册途径。2020年,药品监督管理局及其下属的药品审评中心发布《药品注册管理办法》和《化学药品非处方药上市注册技术指导原则(征求意见稿)》,其中明确了可以直接提出非处方药注册的情形与提交资料要求,这标志着我国药品监管部门对非处方药注册管理规制的放松,然而具体工作流程和规制程度也亟待进一步的探讨。
确立社会性规制均衡理念与准则可有效推进我国非处方药简化注册制度的改革。社会性规制不能一味增强,也不能一味放松,关键是要适度规制,达到规制均衡[4]。因此,本文以美国非处方药专论制度改革为例,分析其如何通过规制目标、规制需求和规制工具的科学设置,实现规制均衡,从而针对我国非处方药专论实施提出适应性建议。
1 社会性规制均衡的内涵
1.1 社会性规制的内涵
社会性规制的定义最早由日本学者植草益提出:“社会性规制是指以保障劳动者和消费者的安全、健康、卫生以及保护环境和防止灾害为目的,对产品和服务的质量以及随之而产生的各种活动制定一定标准,并禁止、限制特定行为的规制[5]。”社会性规制的本质目的是增进人类社会福利,规制内容通常包括环境规制、安全规制和健康规制三种[6]。人类社会总是在一定的环境下生存和发展的,环境规制是社会性规制中的基本内容,主要是政府对大气污染、水污染、噪声污染等负外部性行为的规制,以实现人类可持续发展。安全规制涉及人类的生命保护和财产保护,主要包括劳动安全、交通安全、产品安全等。健康规制关注对人类健康产生威胁的领域,主要包括食品、医药、卫生、废弃物处理等方面的规制[7]。
药品监管领域属于安全规制和健康规制领域,可以从社会性规制均衡视角分析国外先进经验,构建科学的药品监管体系框架。基于非处方药产业同时要求“安全保护”和“促进创新”的特性,本文主要关注在简化非处方药注册途径方面我国可以如何科学规制,不断优化,实现产业技术创新和保护公众健康的均衡。
1.2 社会性规制均衡框架
在药品监管领域,规制和创新一直是被认为矛盾的存在[2]。出于保障健康和安全考虑而制定的社会性规制往往会阻碍创新,但是规制在某些情况下又会为创新创造条件。以往有诸多学者对规制和创新的互动影响进行了研究,主要观点分为促进型、阻碍型和调和型三种:①促进型观点认为良好的规制模式有利于促进基于安全考虑的创新,规避企业激进的创新行为[8],有助于企业在市场需求方面的灵敏反应等;②阻碍型观点认为规制并非有效的产业创新手段,规制成本[9]、过度规制[10]、规制拖延[11]、规制滞后[12]、规制目标不清晰等等都会阻碍创新;③调和型观点认为规制对创新的影响不是促进或阻碍的二元论断,因为不同的规制焦点控制目标会影响产业创新的发展或回退,企业也会根据对政策不确定性的预期或对适应性成本不确定性的预期来调整自身的创新策略。
本文支持调和型观点,认为规制对创新既有促进作用,又有阻碍作用,关键在于如何合理利用规制工具,科学设置规制目标、规制架构、规制标准、规制内容、规制程序等,以实现规制效果的均衡。根据前人研究提出的“目标-供需-工具”的分析框架[2],本文将社会性规制均衡分为目标均衡、供需均衡以及工具均衡:
①社会性规制的目标均衡是实现规制效果均衡的前提。规制的各项设置都是围绕规制目标展开的,总的来说,规制目标越单一、清晰,规制效果越好。因此规制目标均衡研究着力点在于提高各项规制目标间的统一性。对于非处方药产业来说,就在于实现保障安全和促进创新间的平衡;
②社会性规制的供需均衡属于规制的内容层面,主要是评判政府的规制供给与企业的规制需求之间是否达到平衡。若规制供给大于需求,容易引起由于规制过度带来的规制负担,挫伤企业创新积极性,如审评程序繁琐、审评时限过长或不明确等;若规制供给小于需求,则又可能会由于规制缺失导致安全性无法保证。因此需尽量保证规制供给与规制需求的对等;
③社会性规制的工具均衡是实现规制效果均衡的手段。规制工具可分为“激励型规制”和“约束型规制”两类,二者都是为了达到规制目标所采取的一系列措施。在鼓励非处方药的创新发展中,激励型规制包括企业或相关利益相关者与药品监督管理部门之间定期开展沟通会议、创新品种享有独占期、信息及时披露等;约束型规制包括价格控制、制定严格标准、畅通社会监督渠道[13]等。在运用时要注意把控激励和约束的实施强度,最大程度上使两种工具相辅相成,谨防二者的双重弱化。
如图1所示,在理想状态下,规制与创新之间可以通过相互影响与相互制约实现规制均衡。但是在实际情况中,想要达到规制均衡是较为困难的事情,政府需通过实践不断改进规制策略,通过动态调整达到相对均衡的状态。因此本文以美国非处方药专论改革为例,从社会性规制均衡视角剖析其改革背后的逻辑,以期为我国实施非处方药专论提出相应建议。
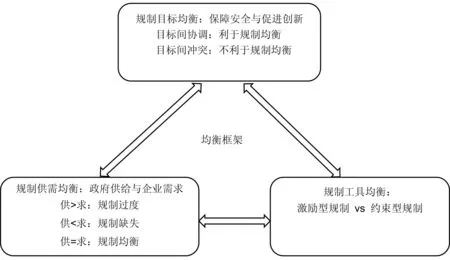
图1 社会性规制均衡框架
2 美国非处方药专论的发展历程与改革内容
2.1 美国非处方药专论的发展历程
2.1.1 美国非处方药专论的提出
非处方药主要用于治疗一些轻微疾病,具有安全性高、质量稳定等特点,消费者不需要医师开具处方便可在网络或零售药店自行购买,主要类别包括感冒药、止痛剂、退烧药等。美国在1938年颁布的《食品、药品和化妆品法》和1962年颁布的《药品修正法》中,规定了药品上市要证明其安全性和有效性,其中也包含非处方药。在此背景下,美国食品药品监督管理局(Food and Drug Administration, FDA)药品评价和研究中心(Center for Drug Evaluation and Research, CDER)下属的OTC评价部于1972年对OTC药品的安全性、有效性、以及标识正确性进行了一次最为全面和系统的评价。由于当时美国市场上所涉及的OTC产品多达35万种[14],如果以每一个产品为对象进行审评,工程量过大,实际操作性较低,因此FDA的审评策略是对这些产品中所含有的700多种活性成分进行评价,并将不同的活性成分按照治疗类别划分,制定若干个适合含有该成分的所有制品的标准。在历经10余年的审评后,FDA制定了含有26类治疗类别的《OTC药品最终专论》,这些专论以成文的形式收录于《联邦法典》中(Code of Federal Regulations,CFR 331-358),具体类别见表1。
目前美国非处方药上市分为新药注册(New Drug Application, NDA)和非处方药专论(OTC Monograph)两个途径。非处方药专论类似于“标准工具书”,用来评判拟上市非处方药是否被认为安全有效(Generally Recognized as Safe and Effective, GRASE)。其评价的是药品成分而非药品个体,若拟上市非处方药成分符合非处方药专论要求,则无需经FDA审批,在符合良好生产规范(Good Manufacture Practice, GMP)的条件下,只需备案即可上市。目前美国市场共有超过10万种非处方药,其中大部分是通过非处方药专论途径上市的[15],由此可以看出美国专论的重要性及必要性。
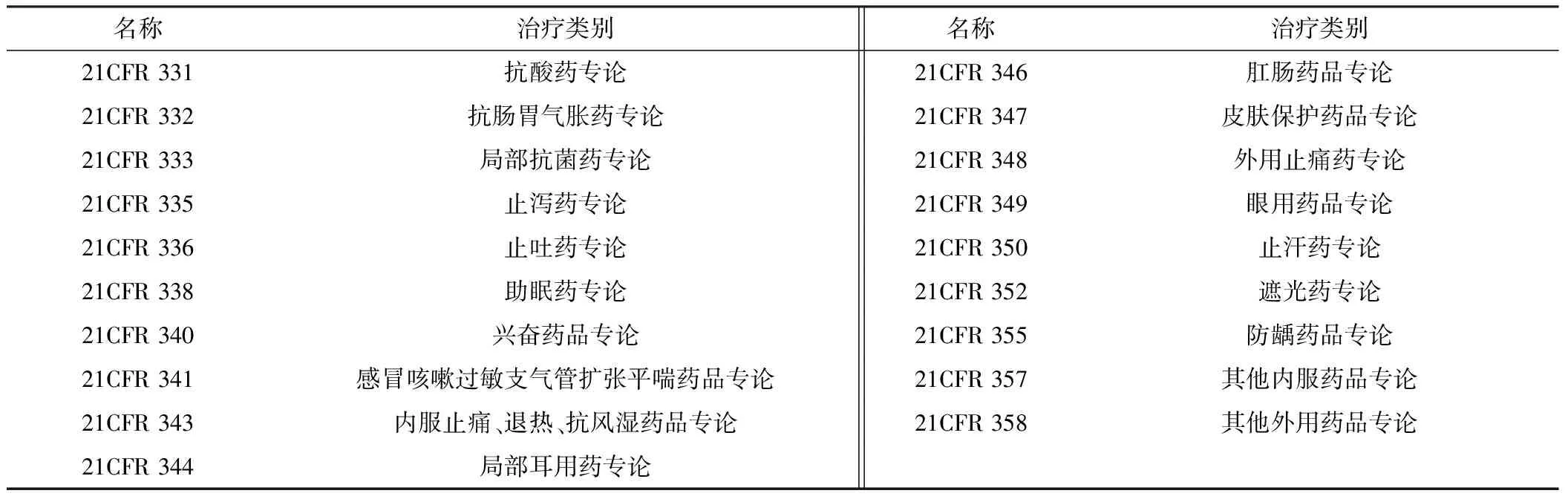
表1 美国非处方药专论类别
2.1.2 美国非处方药专论的改革
自美国1972年建立非处方药专论以来,该途径已成为非处方药在美国上市的主要方式。然而,美国已经实施了将近50年的非处方药专论已无法适应现有非处方药市场的创新需求,存在规制滞后的弊端。多年来,FDA和利益相关者一直在试图改革非处方药的监管。2020年3月19日,美国国会通过了《新冠病毒援助、救济与经济保障法案》(Coronavirus Aid, Relief, and Economic Security Act),简称《CARES法案》,随后美国总统在2020年3月27日正式签署了该法案,标志着非处方药品监管改革的正式启动。此外,美国《食品、药品和化妆品法》第744L和744M条也对专论改革内容增添了相应规定。
《CARES法案》A部分-Ⅲ.F为对非处方药专论管理进行改革的条款,主要包括实施行政令程序以及《非处方药专论使用者付费法案》。改革旨在提高专论修订效率、缩短专论审评时间、促进创新、更快响应紧急安全问题以及为处理非处方药专论活动提供资金等。
2.2 美国非处方药专论改革内容
2.2.1 实施行政令程序(Administrative Order)
非处方药专论包含近800种不同的活性成分,可用于治疗近1400种适应症。然而,从1972年至今的近50年内,药品市场变幻莫测,许多专论内容亟待更新。此前专论的制定与修订审评步骤繁琐,审评时限长,FDA无法及时对非处方药专论进行修改,这导致美国政府无法及时处理公共卫生突发事件。此外,在医药产业迅猛发展及变革的环境下,繁琐的专论构建及修订过程会挫伤非处方药企业的创新积极性,阻碍非处方药产业的进步与发展。
因此美国此次非处方药专论改革提出实施行政令程序,主要对专论的构建与修订过程进行改进,这有助于FDA在保持高标准的同时,更快做出决策。FDA和相关企业(指在《CARES法案》中定义的负责生产、加工、研制或销售药品的个人和集体)都可以发起行政令程序。若发起人是企业,则该请求被称作OTC专论命令申请(OTC Monograph Order Request,OMOR)。
在实施CARES法案之前,非处方药专论制定需经过组建顾问审查组、申请人报送资料和数据、顾问审查小组审评、顾问审查组向局长报告、局长在美国联邦政府公报(Federal Register, FR)上发布“建议制订条例的进一步通告”(Advanced notice of proposed rulemaking,ANPR)、公布“暂行的最终专论稿”(Tentative Final Monograph,TFM)和公布最终专论(Final Monograph,FM)七个步骤;专论修订需要申请人提交公民请愿书或历时及应用范围申请,审评流程繁琐,时限过长。
实施行政令程序后,专论构建与修订程序简化为企业提交OMOR申请、FDA对OMOR进行归档、FDA发布拟定行政令(Proposed Order)、公众评议和FDA公布最终行政令(Final Order)五个步骤[16]。FDA可直接做出对专论内容进行增加、删除以及修改的决定,不需再经过法规规定的完整且繁琐的审批流程,如图2所示。由于移除了多项法规制定所需的批准程序,《CARES法案》特别涵盖了争端解决、听证及司法审议的条款,以确保程序透明公平。
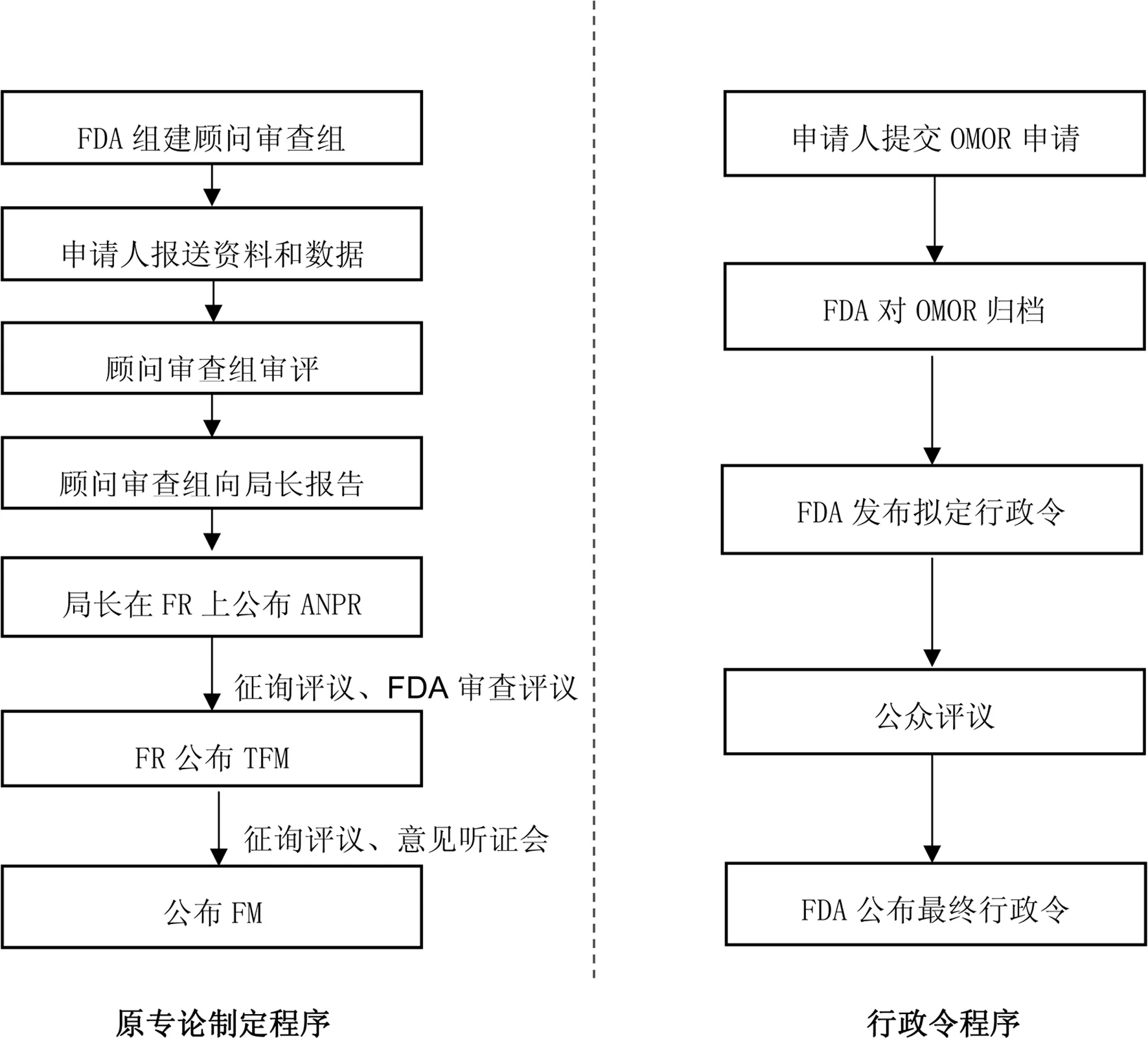
图2 美国非处方药专论改革前后专论制定程序对比
2.2.2 制定《非处方药专论使用者付费法案》(Over-the-Counter Monograph Drug User Fee Program, OMUFA)
目前由于缺乏足够的审评资源,美国专论审评进展缓慢、新的专论无法形成、对安全问题响应不够及时。为解决以上问题,《CARES法案》中提出建立《非处方药专论使用者付费法案》,向企业收取一定费用,为FDA处理非处方药专论提供资金支持。此收费计划旨在为FDA提供额外资源,使其能够在规定时间内高质高效完成专论审查工作,使公众能够获得更多安全、有效、新颖的非处方药。
美国FDA于2021年3月26日公布了《非处方药专论使用者付费法案2021财年费率》。通过OMUFA收取的费用将用来支持FDA在审查场地、提供建议以及审查新的OTC专论命令申请方面的活动[17]。如表2所示,缴纳费用分为两部分:①符合资格的OTC专论药生产场地所有人所缴纳的场地费(Facility Fees);②OTC专论命令申请提交者所缴纳的申请费(OMOR Fees)。
FDA会对专论药场地(Monograph Drug Facility, MDF)和合同加工外包组织场地(Contract Manufacturing Organization, CMO)进行审查并收取相应的场地费。MDF是指生产或加工OTC专论药成品制剂的国外或国内场地,不包括涉及临床研究原料生产、外包装更换及检测的场地;CMO也是非处方药生产机构,但是该场地所有者及其附属的生产场地所有者不会直接向美国批发商、零售商或消费者出售非处方药。FDA将向符合资格的 MDF 场地收取全额设施费($20,322),而对符合资格的 CMO 场地收取三分之二的场地费($13,548)。场地费总额会根据通货膨胀率每年调整,每个企业需要支付的费用由FDA统计的场地总数决定。
此外,FDA还将向提交OTC专论命令申请(OMOR)的企业收取费用。OMOR 分两级,1级(Tier 1)OMORs通常针对在原有专论基础上提出增加新活性成分、新适应症或新药物类别的申请,费用为$500,000;2级(Tier 2)OMORs则针对其它较小的专论变更申请,如调整事实标签顺序、明确特定活性成分浓度或剂量等,费用为$100,000。OMOR是一次性费用,类似于处方药和仿制药收费计划中的申请费,需在提出申请时全部缴清。
OTC专论改革有助于提高审评效率,缩短审评时间,促进非处方药创新。新的行政令程序可提高FDA对非处方药问题的响应速度,提高用药可及性;《非处方药专论使用者付费法案》为FDA专论审查、场地审查以及评估活动提供资金和资源,可有效解决目前停滞不前的专论审评问题。
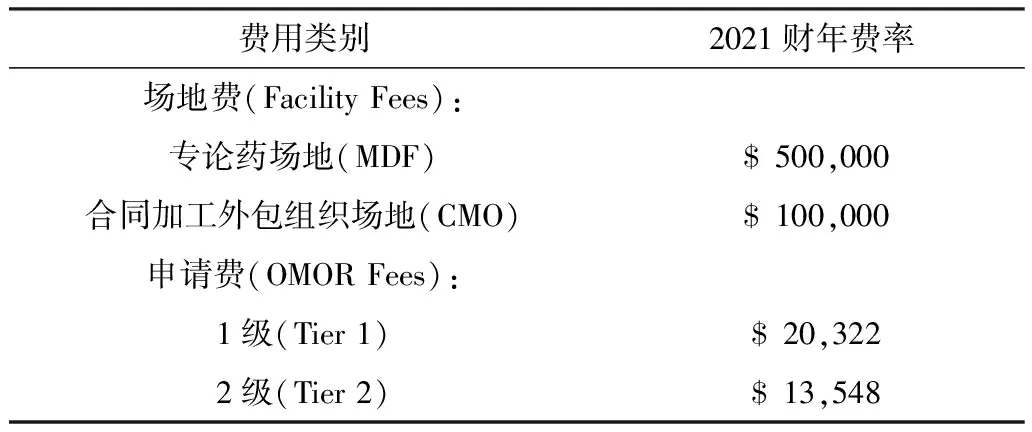
表2 美国非处方药专论2021财年费率
3 美国非处方药专论制度改革特点分析——基于社会性规制均衡视角
政府进行规制的最终目的都是期望达到规制均衡,但这并非能够一蹴而就,而是需要在实践中不断摸索,这也是国家政策不断修正变化的原因。规制均衡的本质是利益均衡,若当前规制不能使政治家、立法者和规制者、产业利益集团和消费者利益集团均处于满意状态,则当前制度必定面临革新。美国非处方药专论改革也是如此。原有的非处方药专论制度已不能与日益发展的非处方药产业相适应,在既有专论体系下,FDA、企业和消费者都无法从中获益,因此改革势在必行。基于前文提出的社会性规制均衡框架,本文将从目标均衡、供需均衡和工具均衡三方面来探讨美国非处方药专论改革如何实现保护安全与促进创新之间的均衡。
3.1 目标均衡——设定协调的规制目标:提升FDA专论处理效率,鼓励企业创新
美国非处方药专论已实施近50年,非处方专论药的安全性已经可以得到保障,因此此次美国专论改革主要针对促进创新设定了清晰且协调的规制目标,即:提升FDA专论处理效率,鼓励企业创新。
规制目标是整个社会性规制框架构建的起点,一切政策、制度以及工具都是以明确的规制目标为导向提出和完善的。美国原有非处方专论制度存在建立和修订步骤繁琐、专论药品审评时限不明确、资源投入有限、缺乏激励措施等弊端,一方面使FDA无法及时有效地对应对公共卫生突发事件,另一方面会挫伤企业研发创新非处方药的积极性。因此在此次专论改革的第一轮周期,FDA对2021-2015年的目标进行总体规划,全面围绕“提升FDA专论处理效率,鼓励企业创新”,进行顶层制度设计及政策工具开发。
2021年为美国非处方药专论改革实施首年,为确保达到总体规制目标,FDA将其拆解成若干个子目标,如在审批人员招聘与培训、IT信息平台建立、申请提交流程、申请者沟通会议、争端解决等方面设定时限要求和完成情况评价。在改革实施前三年(即2021-2023年),FDA工作内容主要聚焦于顶层指导文件的拟定以及审评人员招聘与培训;在改革实施的第四年和第五年(即2024-2025年),FDA的主要工作目标是处理OMOR提交文档、开展申请者沟通会议等。
3.2 供需均衡——简化专论处理程序,明确具体审评时限,降低企业负担
对于美国原有的非处方药专论体系来说,政府处于过度规制的状态,导致企业承担较大的规制负担,不愿提出非处方药创新申请。如在实施CARES法案之前,专论的构建是一个三阶段层层推进的公共规则制定过程,需在美国联邦政府公报上陆续公布《建议制定条例的进一步通告》、《暂定最终专论》,接受公众评议后,才可发布《最终专论》;专论的修订也需经历提交资料、多次公示、多轮评议的过程,程序繁琐,历时较长。
因此此次改革美国政府充分考虑企业的创新需求,简化专论的制定及修订程序,提出行政令程序,且对每个步骤规定了具体时限,在保证药品安全性有效性和信息透明性的前提下,极大地提高了FDA审评效率。FDA对申请较大变更的1级OMOR和申请微小变更的2级OMOR在审评期限上加以区分,避免在较小变更的审评审批上浪费过多的审评资源。每类申请的总审评时限在一年半以内,若拟定行政令发布后收到大量公众评议,1级OMOR的审评时间要延长5个月,2级OMOR的审评时间延长3个月,但总审评时长不会超过两年。各个节点所用时长见表3。
3.3 工具均衡——采取非处方药付费措施,提出会议沟通机制
在非处方药专论改革中,建立的核心政策工具即为《非处方药专论使用者付费法案》,其存在本身对企业来说就有约束性和激励性的双重作用。按照一般逻辑,企业会根据适应性成本与其可能产生的创新收益之间的预期进行比较,做出创新与否的选择。从激励角度来说,拥有专论非处方药制剂生产场地的企业需考虑其每年必须缴纳的场地费,在不进行创新投入的情况下,该费用则变成了沉没成本,无法带来新的创收;从约束角度来说,若企业想要提出创新申请,又需缴纳申请费,这就限制了企业泛滥申请,倒逼企业在提交申请时谨慎考量,做出真正有利于非处方药创新、有利于人民群众利益的申请决定,同时避免审评资源的浪费。

表3 OMOR审评各阶段时限
规制工具服务于规制目标。此次专论改革牢牢把握“提升FDA专论处理效率,鼓励企业创新”的总体目标,制定了一系列的激励性规制工具,其中以开展OMOR申请人沟通会议最为典型。沟通会议旨在增进申请人与FDA的沟通,在申请的各个阶段为申请人提供专业性建议,提高申请通过率。申请人与FDA之间的正式沟通会议分为三类:X类、Y类和Z类。X类会议为紧急必要会议,通常发生于OMOR被拒绝归档或FDA拒绝发布行政令的情况。申请人需要在申请被拒三个月内提出X类会议申请,以确保后续工作的持续推进。Y类会议常常由申请人在专论OMOR申请过程中的重要节点提出,目的是帮助申请人明确数据提交要求,及时获得FDA反馈并改正以提高申请成功率,主要包括数据要求会议(Overall Data Requirements Meetings)、提交OMOR前会议(Presubmission Meetings)和审评中会议(In-review Meetings),其各阶段会议具体沟通事项见表4。Z类会议为其他任何必要性会议。
除沟通会议外,其他激励措施还包括增加专论新活性成分或适应症的企业享有18个月的市场独占期、IT信息化平台的建立、信息公示透明化、FDA对拟处理专论内容进行公示等。这些激励工具都有助于增进企业投入研发、提出申请的积极性。美国非处方药专论改革后开发的种种激励性规制工具与规制目标匹配程度较高,决定了其公共规制的高质量水平。

表4 各阶段Y类会议沟通事项
4 美国非处方药专论改革对我国实施非处方药专论的启示
4.1 基于风险管理理念,探索建立非处方药简化注册路径
目前我们国家尚未建立非处方药专论制度,因此若想实施类似于美国非处方药专论制度的非处方药简化注册路径,首先需要设定清晰、明确且尽量单一的目标——即在保证药品安全、有效、质量可控的前提下,尽可能简化注册渠道,加快非处方药的上市。在此可以引入风险管理理念,将专论建立过程作为风险管控过程,从制定风险管理计划、风险识别、风险评价、风险控制和风险沟通五个环节有效降低药品全生命周期的安全性风险。
通过对美国非处方药专论的研究,本文认为我国非处方药专论建立过程可分为以下七个步骤:(1)组建OTC审评专家小组;(2)确定专论药品治疗领域,制定不同类别OTC药品候选活性成分筛选标准;(3)确定入选OTC药品活性成分的安全性、有效性评价标准;(4)收集OTC药品活性成分评价材料(企业递交或主动审查);(5)对所收集的活性成分进行安全性、有效性评价;(6)形成初步的“活性成分标准”;(7)公布“活性成分标准”与公开征求意见并确定终稿专论。
4.2 关注企业诉求,完善配套政策体系,引导企业积极参与创新
非处方药企业方纷纷表示行业政策和企业政策是相辅相成、互促共进的,非处方药行业发展需要良好的环境进行开放式创新[18]。因此我国若要实施非处方药专论,首先应在法律层面上做出硬性规定,同时提出各项可以指导企业具体实践的配套政策并积极落实,充分发挥政策体系在领域中的管理、协调、评价和监控等规制作用[19]。
如美国在专论改革方面,首先出台了《CARES法案》,标志着非处方药专论改革的正式启动。随后立即实施《非处方药专论使用者付费法案》,并在《联邦公告》(Federal Register, FR)上公布了《非处方药专论使用者付费法案2021财年费率》,及时规范企业行为。美国还发布了《2021-2025财年非处方药专论使用者付费计划绩效目标和程序》,制定了改革目标及实施细则。除此之外,为引导企业创新,助力改革推进,美国计划五年内发布一系列指导文件,如《会议指导文件》、《电子化提交指导文件》、《争端解决指导文件》等[20]。我国也可借鉴美国经验,从顶层制度设计出发,建立非处方药专论制度,并细化落实各项针对企业具体的实施细则,有助于更多非处方药的上市流通。
4.3 逐步放开非处方药管理力度,加强激励工具的开发
我国是全球非处方药管理最严格的国家之一,一款药品从申请到审批通过通常需要五年时间,上市速度几乎和非处方药相同。2020年以前,我国处方药和非处方药采用同样的审批流程及技术要求。2020年1月15日年通过的《药品注册管理办法》规定满足特定条件的非处方药可以制剂提出上市许可申请,这是我国自从2000年开始实施《处方药和非处方药分类管理办法(试行)》以来,对OTC审评审批做出的较大变化,说明国家政策向放开非处方药管制,加快非处方药上市倾斜。
在制度放开基础上,创新非处方药是否能够真正走入市场更多取决于企业是否有意愿提交非处方药注册申请。因此在建立非处方药专论制度时,可以借鉴美国非处方药专论改革中采用的激励工具,如增强申请者与CDE间的沟通,设定沟通节点,明确沟通事项,以提高非处方药专论申请的成功概率,激励更多企业提出非处方药申请。除此之外,CDE还可以通过定期在官网上公布近期重点关注治疗领域或药物品种、赋予首次申请创新非处方药企业独占期、建立电子化资料提交平台等,提高审评效率,鼓励企业创新。
猜你喜欢
教育家(2022年17期)2022-04-23
房地产导刊(2022年4期)2022-04-19
意林(2021年3期)2021-03-11
父母必读(2021年3期)2021-02-04
汉语世界(The World of Chinese)(2020年6期)2020-12-21
健康之家(2019年9期)2019-12-14
大经贸(2019年1期)2019-04-26
新经济导刊(2018年10期)2018-10-18
民生周刊(2018年10期)2018-06-07
职工法律天地·下半月(2017年12期)2018-02-26
