
氧化铜表面臭氧分解路径及表面氧物种生成机理研究
2022-08-13 06:56邵广才王广钊王军锋
分子催化 2022年3期
龚 朋, 刘 璐, 邵广才, 王广钊, 王军锋*
(1. 江苏大学 能源与动力工程学院, 江苏 镇江212013;2. 长江师范学院 电子信息工程学院 超常配位键工程与新材料技术重庆市重点实验室, 重庆 408100)
臭氧(O3)的催化分解是环境污染控制领域的重要反应. 一方面O3本身是一种污染物, 会导致光化学烟雾, 影响人体肺功能, 引起中枢神经障碍[1-2],而催化分解是去除O3的有效方法[3-7]之一. 另一方面, 在催化O3深度氧化和等离子体-催化技术降解有机物过程中, 高压放电产生的O3在催化剂表面解离成氧化能力更强的活性O原子和OH自由基, 从而对反应物和放电过程中未被完全降解的产物进行深度氧化, 达到去除有机物的目的[8-9]. 近年来,人们对催化分解O3反应进行了广泛研究, 过渡金属氧化物由于成本低、 毒性小等优点, 是进行O3分解的重要催化剂之一, 逐渐成为研究热点[10-11]. 氧化铜(CuO)广泛应用于催化O3分解过程, 例如SiO2负载金属氧化物分解O3的效率依次为: Cu/SiO2> Co/SiO2> Ni/SiO2> Fe/SiO2> Mn/SiO2[12]. CuO、 CuO/沸石等催化剂能够有效催化O3氧化空气中的甲醛[13-14].Cu2O-CuO-Cu(OH)2分级纳米结构在室温下表现出100%的O3分解效率, 并具有高稳定性和抗水性, 纳米结构的表面形态和化学成分在催化O3分解中起着重要作用, 纳米结构中的离域空穴能促进中间产物氧阴离子和催化剂表面之间的电子转移[15].


1 计算方法及模型
计算采用VASP软件包[22-23], 基于广义梯度近似(GGA-PBE)[24]和投影增强波(PAW)[25]方法, 截断动能为520 eV. 优化后的CuO的晶格参数为a=0.47 nm、b=0.34 nm、c=0.51 nm和β=99.54°,与之前的计算结果基本一致[26]. 根据以往的研究,CuO(111)表面具有较低的表面能, 是最稳定的表面[27]. 采用p(4×2)超胞建模, 三层O-Cu-O结构, z方向上增加1.5 nm的真空层[28-29].
在GGA+U计算中, 设置Cu的Hubbard参数U=7.5 eV, 可以很好地处理Cu 3d轨道的强电子相关性[30]. 表面结构弛豫收敛的判定标准为Hellmann-Feynman力小于0.2 eV/nm[27,31]. 分别使用2×2×1和7×7×1 的Monkhorst-Pack网格进行弛豫和态密度计算. 采用CI-NEB方法搜索过渡态[32].
吸附能(Eads, kJ/mol)和差分电荷密度(CDD,Δρ, 1000 e/(nm)3)的计算方法如下:

其中,EO3+CuO是吸附O3后系统的总能,ECuO和EO3分别为CuO和气相O3分子的总能.

其中,ρO3+CuO是吸附O3后系统的电荷密度;ρCuO和ρO3分别为CuO和O3分子的电荷密度.
2 结果和讨论
2.1 O3在洁净表面的吸附
O3在CuO(111)的表面吸附形成4 种比较稳定的O3构型, 如图1(a)所示. 表1给出了O3在CuO(111)表面的吸附能. 当O3的两端的O原子分别与CuO表面的两个Cu原子成键形成桥式结构时, 吸附能较大, 构型br(i1)和br(i2)的吸附能分别为-0.77和-1.22 eV. 当O3一端的O原子为自由端时, 吸附能较小, 构型top(1)和top(2)的吸附能分别为-0.75和-0.28 eV.br(i1)中O3的O-O键分别为0.13 和0.14 nm, top(1)中的O-O键分别为0.13和0.14 nm, 此时O3趋于分解成O*和O2*; 而br(i2)中O3的O-O键由0.13对称地拉长至0.14 nm, 说明O3的O和表面的Cu具有较强的相互作用, 形成的Cu-O键长较短(均为0.20 nm), 因此该构型的吸附能最大.
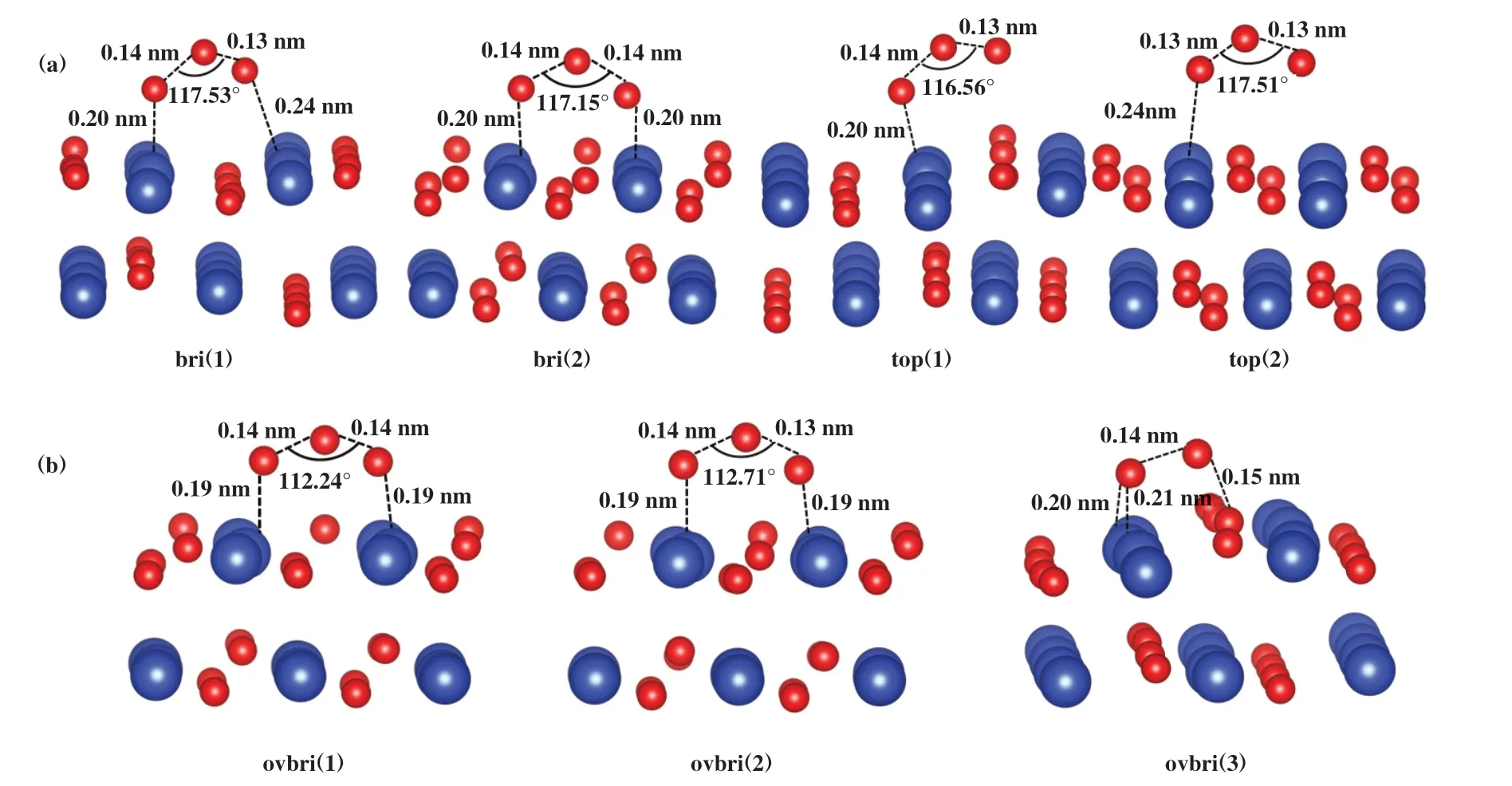
图1 O3吸附在CuO(111)表面的结构侧视图Fig.1 Side view of the configurations of O3 adsorbed on CuO(111) surface

表1 O3在CuO(111)表面的吸附能Table 1 Adsorption energy of O3 on CuO(111) surface
为进一步分析成键情况, 计算了构型bri(2)的差分电荷密度(CDD)图. 如图2(a)所示, Cu吸附位的电荷密度明显减小, O3末端两个O-Cu原子下方的电荷密度显著增强, 说明形成了Cu-O离子键.O3中间的O-O附近电荷密度也有所增强, 说明电荷从CuO表面转移到O3. O3的3个O原子之间的区域电荷密度减小, 表示O3中的O-O键减弱, 与OO键长增长相符. 图2(b)所示为构型bri(2)中O3末端和O3中间O原子的投影态密度(PDOS)图,s轨道和p轨道分别用实线和虚线表示. O-Cu和O-O的s轨道均在-14 eV处有强峰, 在-7 eV处有弱峰;p轨道在-8.56、 -8.31、 -6.82、 -6.57、 -0.58 和-0.09 eV处交叠, 说明O-Cu和O-O之间仍有很强的共价键. 由于Cu-O键的形成, 在-2.70、 -1.83、 -1.59和-1.34 eV处有强峰, 而O-O在该处无峰, 说明O-Cu和Cu形成了很强的离子键.

图2 (a) 构型bri(2)和ovbri(1)的CDD图; (b) 构型bri(2)和(c) ovbri(1)的投影态密度(黄色和蓝色部分分别表示电荷密度增加和减少了50 e/(nm)3; E=0 eV为费米能级;O-Cu表示O3中与Cu成键的O, O-O表示O3中间的O)Fig.2 (a) CDD plotof bri (2) and ovbri (1) with an isosurface level of 50 e/(nm)3; charge accumulation is in yellow and charge depletion is in blue. PDOS of O-Cu and O-O in (b) bri (2) and (c) ovbri (1), where O-Cu represents the O at one edge of O3 that bound with Cu and O-O represents the O in the middle of O3. The energy at E= 0 eV represents the Fermi energy.
2.2 O3在具有氧空位的CuO(111)表面的吸附
氧空位是金属氧化物表面普遍存在的一种表面缺陷, 在氧化还原反应中起重要作用, 因此, 研究了O3在具有氧空位的CuO表面的吸附. 图1(b)是O3在含有氧空位的CuO(111)表面Cu位上的3种吸附构型ovbri(1)、 ovbri(2)和ovbri(3), 均为桥式构型, 不再形成O3一端的O原子为自由端的构型. O3在含有氧空位的CuO(111)表面的吸附能远高于在完整CuO(111)表面的吸附能, Cu-O键长变短, 说明Cu和O-Cu的相互作用变强. 其中ovbri(3)吸附结构的吸附能最大, 为-2.95 eV, O3一端的O原子填补了氧空位,另一端的O原子与表面两个Cu形成桥键, O-O键分别拉长至0.14和0.15 nm.
由图2(a)构型ovbri(1)的差分电荷密度图可以看出, 与完整CuO表面的O3吸附相比, O3在具有氧空位的CuO表面吸附时, 电荷转移明显增强, Cu附近的电荷密度减弱程度和O3的O原子附近电荷密度增强程度均大于完整表面, 说明O3分子和CuO表面存在强烈的相互作用. 从图2(c)构型ovbri(1)的PDOS图可以看到, O-Cu和O-O在费米能级附近的态密度减弱并向低能量偏移, O-Cu和O-O的p轨道在-8.63、 -6.90、 -0.52 和-0.22 eV处交叠, 能量低于完整表面, 说明O3在具有氧空位的表面吸附的结构更稳定.
2.3 O3在CuO表面的分解路径和表面氧物种的生成
反应路径终态吸附的O2*和O*是能够引发氧化反应的表面活性氧, 表2列出了这些表面活性氧的价态和键长. 吸附的O*价态为-0.55和-1.61; 完整表面吸附的O2*价态分别为-0.04、 -0.21、 -0.18和-0.15, O-O键长分别为0.12、 0.13、 0.13 和0.13 nm, 略高于自由氧分子的O-O键长(0.12 nm), 氧空位处吸附的O2*价态为-0.10和-1.03, O-O键长为0.12和0.15 nm, 可见O3分解后形成的表面吸附O2*和O*外层电子配位均不饱和, 形成了超氧物种[33],相对于自由O2更容易参与氧化反应.
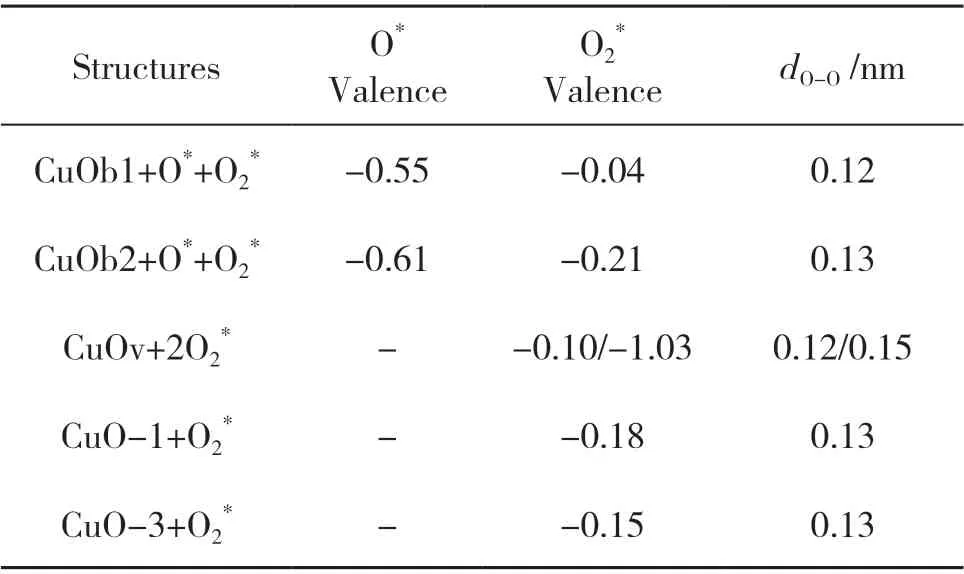
表2 反应路径终态表面活性氧的价态和键长Table 2 Valence states and bondlengths of surface active oxygen in the final state of reaction path
根据O3在CuO催化剂上吸附的初始结构, 提出构型br(i1)、 br(i2)、 ovbr(i2)和ovbr(i3)分解并形成表面活性氧的合理途径. O3在CuO(111)表面分解的活化能垒图, 过渡态和终态的几何结构如图3所示. 对于O3在完整CuO(111)表面的分解, 反应从构型br(i1)和br(i2)开始, O3两端的O原子与CuO表面的两个Cu原子形成桥式结构, 随着O3的O原子与CuO表面的Cu原子相互作用, 电子从Cu原子转移到O原子上,O3与CuO结合形成中间体, 即过渡态TS1和TS2, 最终O3解离成O*和O2*, 反应的最高能垒分别为0.58(路径bir(1))和0.52 eV(路径bir(2)). O2*是弱吸附分子, 从表面脱附分别需要0.42 (路径bir(1))和0.52 eV(路径bir(2))的能量, 脱附后形成自由氧分子. O*和CuO表面的晶格氧结合, 形成另一个O2*, 此O2*脱附需要较高的能量2.06 eV, 脱附后形成含有氧空位的表面. 该过程中, 氧空位形成的能垒最高; 若不考虑氧空位的形成, 以bir(2)为初始结构的反应路径能垒较低, 为0.52 eV, 是反应可能发生的路径.

图3 O3在CuO表面的分解路径和活化能垒图Fig.3 O3 decomposition path and activation energy barrier on CuO surface
O3在具有氧空位表面的分解从构型ovbri(1)和ovbir(3)开始, 在ovbir(1)中, O3中的两个O与表面的两个Cu分别成键, 形成较为对称的桥式结构, 吸附能为-1.66 eV. 在ovbir(3)中, O3一端的O原子与CuO表面的两个Cu原子形成桥式结构, 另一端的O原子填补了氧空位, 吸附能为2.95 eV. 吸附后的O3经TSl和TS2两个过渡态形成O2*, 另一个O变成CuO表面的晶格氧, 反应能垒分别为0.30 (路径ovbir(1))和0.12 eV(路径ovbir(3)), O2*从反应表面脱附后, 留下完整的CuO表面, 脱附所需能量分别为0.27 (路径ovbir(1))和0.52 eV(路径ovbir(3)). 由此可见, 以ovbir(3)为初始结构的反应路径能垒较低, 是反应可能发生的路径, 但是该路径最终O2*脱附的能垒比ovbir(3)路径高. 同时, 在具有氧空位表面, O3分解的反应能垒低于完整表面. 氧空位的形成提高了吸附能, 使O3分子更容易吸附在CuO表面, 从而促使催化反应发生; 同时, 氧空位的形成降低了反应能垒, 利于反应进行, 因此在具有氧空位的表面O3更易发生分解.
3 结论
采用GGA+U计算详细探讨了O3在CuO(111)表面的吸附和解离过程. O3分子在完整CuO表面吸附形成4种稳定构型, 3种构型为化学吸附, 吸附能最高为-1.22 eV, 1种构型为物理吸附, 吸附能为-0.22 eV. O3在具有氧空位的CuO表面吸附形成3种稳定构型, 均为化学吸附, 吸附能最高为-2.95 eV, 显著高于完整表面的吸附能. O3吸附后, Cu吸附位的电荷密度减小, O3中的O原子附近的电荷密度显著增强, 电荷从CuO表面转移到O3, 并形成Cu-O离子键. O3在完整CuO(111)表面的分解从桥式构型br(i1)和br(i2)开始, 通过反应能垒分别为0.58和0.52 eV的过渡态, 解离成O*和O2*, O2*的脱附能分别为0.42和0.52 eV. O*和CuO表面的晶格氧结合形成的O2*脱附后形成氧空位, 脱附能为2.06 eV. O3在具有氧空位表面的反应从构型ovbr(i1)和ovbi(r3)开始, 经反应能垒分别为0.30和0.12 eV的过渡态解离形成O*和O2*, O*变成CuO表面的晶格氧, O2*从CuO表面脱附后形成完整CuO表面, 脱附能分别为0.27和0.52 eV. 氧空位的形成增大了吸附能, 降低了反应能垒,使O3分子更容易吸附在CuO表面, 加快了O3的催化分解. O3分解后形成了超氧物种, 提高了表面的氧化活性.
猜你喜欢
航空动力(2022年2期)2022-05-06
小作家报·教研博览(2022年11期)2022-04-02
安徽工业大学学报(自然科学版)(2021年3期)2021-09-08
中学课程辅导·高考版(2020年9期)2020-10-20
校园英语·月末(2019年11期)2019-09-10
分析化学(2018年7期)2018-09-17
新高考·高一物理(2016年7期)2017-01-23
新高考·高一物理(2015年6期)2015-09-28
新高考·高一物理(2015年6期)2015-09-28
读者欣赏(2014年6期)2014-07-03
