
多发性骨髓瘤合并JAK2阳性原发性骨髓纤维化1例报告并文献复习
2022-08-24 01:27马家乐魏威訾杰马金龙葛峥
实用老年医学 2022年8期
马家乐 魏威 訾杰 马金龙 葛峥
多发性骨髓瘤(MM)是以浆细胞及其前体在骨髓中异常增殖为特征的恶性肿瘤,起源于B细胞生发中心,并具有分化为浆细胞的能力[1]。原发性骨髓纤维化 (PMF) 是一种Ph染色体阴性骨髓增殖性肿瘤(MPN),起源于可分化的多能骨髓造血干细胞[2]。临床上MM合并骨髓纤维化(MF)的病人并不少见,但多数为继发性MF(SMF),其发生率为 20.5%[3]。SMF的存在与 MM 病人的浆细胞浸润程度、较高的骨髓瘤 ISS 分期和较差的预后有关[4]。但是MM与PMF的关系目前仍存在争议,而且国内鲜有报道。现报道1例MM合并PMF病例,并复习相关文献。
1 病例资料
病人女,73岁,因“乏力、心悸、头痛1个月”就诊于当地医院。查血常规:WBC为6×109/L,Hb为75 g/L,PLT为114×109/L,β2 微球蛋白(β2-MG)为2.5 mg/L,乳酸脱氢酶(LDH)为313 U/L,CRP为50 mg/L,球蛋白为42 g/L;血清蛋白电泳检测:M蛋白为15.4%;免疫固定电泳:发现IgGλ型M蛋白;外周血流式:见5.3%的髓系抗原表达的幼稚细胞群体,7.3%的嗜碱性粒细胞群体。外周血二代测序(NGS):检测到JAK2 p.(V617F)突变率为48.20%、TP53 p.(Y234C)为17.30%、TP53 p.(Y220C)为21.90%,考虑浆细胞病,未予治疗。后于2020-10-14入住中大医院血液科。既往有高血压和糖尿病病史,无毒物、药物接触史,个人史、家族史无特殊。入院查体面色苍白,无肝脾及淋巴结肿大。
辅助检查:WBC为6.16×109/L,Hb为67 g/L,PLT为115×109/L;肝肾功能正常;钙为1.98 mmol/L,白蛋白为31.9 g/L,LDH为262 U/L;β2-MG为2.58 mg/L;免疫全项:IgG为17.2 g/L,LAM轻链为4.71 g/L;血清免疫固定电泳:单克隆IgG-λ阳性;血清蛋白电泳:3.23 g/L;尿免疫固定电泳示:单克隆免疫球蛋白IgG-λ;24 h尿蛋白为0.332 g/24h;血沉为105 mm/h。骨髓“干抽”,涂片示增生减低,原始粒细胞占 2%(图1A)。重复骨穿结果大致同前(图1B、1C)。外周血偶见原始细胞和有核红细胞。骨髓活检显示明显纤维化(3级),可见病态巨核细胞和可疑巨核细胞簇,浆细胞增多(占30%)。免疫组化:浆细胞数量增加,最高计数约占30%,以Lambda单克隆表达为主。免疫组化结果:CD117(个别阳性),CD235a(红系+),CD34(个别阳性),CD61(巨核+),MPO(粒系+),CD138(+,约占30%),CD38(个别阳性),Kappa(-),Lambda(+),Mum-1(+,约占20%)。特殊染色结果:铁染色(-),网状纤维染色(3+)。流式免疫分型示:CD34+CD117+CD33+CD38+CD7-HLA-DR+细胞占2.84%。荧光原位杂交技术检测(FISH):检出5q-(31%)、7q-(8%);未检出:17p-/-17、20q-、-7;BCR-ABL1/ABL1融合基因阴性。基因突变:JAK2 p.(V617F)为54.11%、JAK2 p.(L808W)为74.54%、CBL p.(I393N)为3.27%错义突变、TP53 p.(Y234C)为24.71%、TP53 p.(Y220C)为20.76%。染色体:44~46,XX,-5,-9,-13,+mar,+r,inc[cp8]/46,XX[12]。正电子发射计算机断层置像(PET-CT):全身广泛骨质改变,符合MM;部分椎体稍压缩改变;双小腿周围肌群水肿、萎缩;双侧踝关节、膝关节腔少量积液;肝及双肾小囊肿;脾大,脾脏小血管瘤可能。双下肢血管彩超示:双下肢动脉粥样硬化(斑块形成),深静脉血流通畅。腹盆部CT平扫示:肝囊肿;肝内钙化灶;脾大;左肾微小结石;盆底脂肪间隙钙化灶;盆腔少量积液;动脉粥样硬化;多发骨质改变,骨髓瘤?头颈部磁共振血管造影、脑电图均未见异常。

图1 患者不同阶段骨髓形态学变化
诊断:(1)MM IgG-λ(DS分期Ⅲ期A组,ISS分期Ⅱ期);(2)PMF;(3)高血压;(4)2型糖尿病。
治疗及转归:分别于2020-10-22、2020-11-27予PCD方案(硼替佐米1.3 mg/m2第1、4、8、11天+地塞米松10 mg 第1~2,4~5,8~9,11~12天+环磷酰胺300 mg 第1,4,8,11天)化疗2个疗程,病人化疗耐受性可,疾病评估达部分缓解,但临床症状无明显改善。在病人确诊2个月后,2020-12-30入院体检发现可触及脾肿大,约肋下2 cm,骨髓涂片提示骨髓增生活跃,粒系可见中毒颗粒,嗜酸性粒细胞和嗜碱性粒细胞略有增加(图1D)。NGS示JAK2 p.(V617F)基因突变率为76.84%、TP53 p.(Y234C)为35.35%和 TP53 p.(Y220C)为34.14%,各基因突变比例增加。于2021-01-01予VRD(硼替佐米 1.3 mg/m2第1、4、8、11天;来那度胺 25 mg 第1~21天和地塞米松 10 mg第1~2、4~5、8~9、11~12天)。化疗后病人骨痛得到短期缓解。但PLT持续下降,低于50×109/L。同时,病人于2021年2月逐渐出现活动耐力下降,双下肢水肿,脾大,约肋下4 cm。骨髓仍“干抽”,涂片示增生减少,原始粒细胞占1.6%(图 1E)。骨髓活检示巨核细胞约0~6个/高倍镜视野,可见形态异常,未见明显浆细胞数量增加,纤维化明显(3级),造血细胞减少,粒细胞和嗜酸性粒细胞增加(图2B)。流式免疫分型示:CD34+CD117+CD33+HA-DR+CD38+CD56-细胞占2.77%。基因突变:JAK2 p.(V617F)为82.12%、JAK2 p.(L808W)为74.54%、CBL p.(I393N)为2.19%错义突变、TP53 p.(Y234C)为37.19%、TP53 p.(Y220C)为36.79%。病人PMF诊断明确,但因PLT水平低,靶向治疗出血风险大,予TD方案(沙利度胺50 mg/d,泼尼松20 mg/d)治疗。2021-02-25病人出现口眼歪斜、左侧肢体无力、骨痛加剧,同时合并严重肺部感染、心功能不全,最终于2021-03-28即MM合并PMF诊断5个月后死于呼吸衰竭。
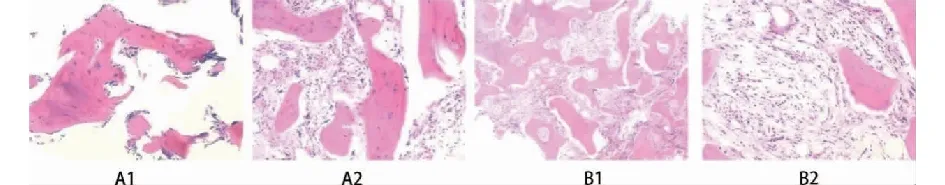
图2 病人不同阶段骨髓活检变化
2 讨论
MM与PMF源自不同的造血克隆,两者共存的情况极为罕见,且两种疾病之间的相互关系尚不清楚。先前的文献中也有报道过MPN合并MM的病例,但多数为SMF[5-12]。对于MM合并SMF或PMF区别在以下几个方面:第一,病因上,SMF通常继发于慢性粒细胞白血病、MM、MPN转化等,而PMF原因不明;第二,诊断及治疗上,MM合并SMF的特点为MF程度相较于PMF低,经治疗后纤维组织增生程度有所降低,而PMF Gomori染色多为强阳性,改善微循环治疗后,纤维化程度缓解不显著[13]。国内外对于JAK2阳性PMF合并MM的报道极其罕见。两种血液肿瘤发生于同一例病人在理论上和临床上都是至关重要的问题。
在本例报道中,该病人初诊时骨髓活检免疫组化提示浆细胞占30%,血清和尿单克隆M蛋白阳性,且存在贫血、多部位溶骨性破坏(PET-CT确认)等靶器官损害的证据,因此病人MM诊断成立。同时,病人初诊时骨髓干抽、骨髓活检见病态巨核细胞和可疑巨核细胞簇,流式细胞术提示原始细胞占2.84%、网状纤维染色(3+),基因突变提示JAK2突变阳性,同时伴LDH升高、血象异常,考虑同时存在PMF。因活动性MM治疗指征明确,经以硼替佐米为基础的方案治疗后,MM达PR,但病人MF的特征,如WBC增多及脾大更加突出,因此更加印证了PMF的诊断。
目前对于MF和MM共存的假说包括:由淋巴因子分泌引起的与MF相关的浆细胞病,即MF克隆继发于MM;两种不同克隆性疾病共存[5];同一种克隆起源,即克隆演化,但并排独立发展[6]。
恶性细胞分泌的淋巴因子可能会刺激肿瘤相关MF的产生,而MM相关的MF是可逆的。Stevenson等[8]报道了1例患有特发性骨髓纤维化(IMF)的MM 病例,他们使用 X 连锁多态性分析骨髓谱系细胞的克隆衍生,证明骨髓增殖性疾病中的骨髓成纤维细胞不是克隆性的,而是继发于分子水平上恶性浆细胞的克隆性增殖。Kawauch等[12]认为在MM相关的MF的发病机制中,来源于骨髓瘤细胞的IL-6等细胞因子刺激骨髓中血小板来源的生长因子(PDGF)等成纤维细胞增殖因子的分泌,促进了纤维化的发生。此外,其他共同的诱发因素,包括骨髓的炎症微环境、烷化或放射性疗法的暴露、慢性炎症和获得性体细胞突变,也在这两种恶性肿瘤的发展中发挥关键作用[14-16]。相对的,在MPNs,与一般人群相比,继发性MM的风险比为1.55 (95%CI:1.0~2.32)[14]。然而,其确切的机制仍不清楚。因此,在诊断时需要排除药物性、骨髓微环境变化对两种疾病发生发展的影响。
在大多数情况下,两种不同的肿瘤共存是否反映了双克隆性仍不清楚,即源于造血过程中2个独立的细胞谱系,或者是由于单克隆肿瘤细胞以不同方式分化的能力[17]。Kuroda等[18]报道了1例MM和原发性血小板增多症(ET)共存的病例,该病例起源于不同分化水平的不同恶性克隆,独立获得不同的分子畸变;他们发现 JAK2V617F 存在于非骨髓瘤外周血白细胞和骨髓造血细胞中,但不存在于来自骨髓的CD138阳性骨髓瘤细胞中。这表明MM和ET来自不同的克隆。这一发现有助于解释两种不同细胞谱系共存的现象。
尽管迄今为止,文献中没有证据支持MPN的淋巴母细胞转化,但不能排除淋巴祖细胞和MPN克隆有同一种克隆起源的可能性。基于这一假设,大多数作者支持单克隆肿瘤细胞双系分化的假设。MM中MPN的发展可以解释为单个祖细胞向髓细胞和浆细胞恶性肿瘤的肿瘤转化。Wang等[19]从MF小鼠的骨髓和脾脏中回收的骨髓和淋巴细胞中均发现JAK2 p.(V617F)突变,表明该突变最初发生在普通造血干细胞 (HSC) 中。与此一致,在CD34+、CD38-HSC和CD34+、CD38+祖细胞群中可检测到JAK2和CALR突变体[20]。有理由怀疑,MM与PMF存在同一克隆起源并发生了克隆演化,且在不同的时间表现出不同的优势。尤其在本病例中,在MM诊断的同时确诊PMF,在MM治疗好转后,PMF的特征更加突出,但至于这一假设是否成立,尚需要更多的研究加以验证。
总而言之,在MM的诊断中需要警惕MPN克隆的存在,尤其是骨髓活检提示存在MF时,需要完善分子学检查进一步排除PMF的诊断,MM病人本身的骨髓微环境变化及治疗期间其他用药情况,也需要引起重视。
猜你喜欢
当代医药论丛(2022年4期)2022-11-26
首都食品与医药(2022年19期)2022-11-19
中国农业科学(2022年16期)2022-09-19
中国临床医学(2022年3期)2022-07-08
临床超声医学杂志(2022年4期)2022-05-07
现代检验医学杂志(2022年2期)2022-04-19
健康体检与管理(2022年2期)2022-04-15
现代临床医学(2022年1期)2022-02-12
中国药学药品知识仓库(2021年18期)2021-02-28
健康必读·下旬刊(2020年10期)2020-10-28
