
Drp1在糖尿病心肌病中的作用机制研究进展
2022-10-16 08:17宋小刚吴冰陈永清
解放军医学杂志 2022年9期
宋小刚,吴冰,陈永清
1兰州大学第二临床医学院,甘肃 兰州 730030;2解放军联勤保障部队第940医院老年科,甘肃 兰州 730050;3甘肃省中心医院心内科,甘肃 兰州 730070
目前,全球糖尿病患者约4.51亿,预计到2045年将增加至6.93亿[1]。超过50%的糖尿病患者死亡或致残的原因为心血管并发症[2]。糖尿病心肌病(diabetic cardiomyopathy,DCM)是一种独立于冠状动脉疾病、高血压和心脏瓣膜病而发生心力衰竭的病理生理状态,早期即可出现心肌纤维化、心肌重塑和舒张功能障碍,随后出现收缩功能障碍,最终发展为心力衰竭[3]。DCM的发生涉及多种机制,包括心脏胰岛素信号传导异常、线粒体功能障碍、氧化应激、炎症反应、钙超载、内质网应激、微血管功能障碍和肾素-血管紧张素-醛固酮系统(RAAS)的激活等[4-5],其中线粒体功能障碍和氧化应激被认为是DCM发生的主要病理生理机制[6-7]。线粒体是高度动态的细胞器,其分裂与融合平衡失调所致的线粒体功能障碍是许多心血管疾病的始动因素,如内皮功能障碍、心肌肥大、心肌病、心力衰竭、心肌缺血再灌注损伤等[8]。动力相关蛋白1(dynamin related protein 1,Drp1)是线粒体分裂的调节因子,在线粒体功能障碍和线粒体氧化应激中发挥着至关重要的作用。本文围绕Drp1在线粒体分裂及DCM中的作用机制进行综述,旨在为DCM的基础研究和临床诊疗提供新思路。
1 线粒体结构
线粒体位于细胞质中,由高渗透的外膜和低渗透的内膜组成,膜间隙含有细胞色素C等可溶性酶及半胱氨酸蛋白酶(caspase)-3、caspase-9的前体和辅助因子。内膜的生物氧化过程可产生跨膜电子梯度,并介导电子传输及磷酸化,从而生成三磷酸腺苷(ATP)。基质中包含线粒体脱氧核糖核酸(DNA)、核糖核酸(RNA)、核糖体、酶等(图1A),是进行三羧酸循环的重要场所。
正常心肌细胞线粒体呈管状、棒状结构,横截面呈椭圆形,长宽比约1.5,密集地排列在心肌纤维间隙中,平均长度约7.97 μm,而过度分裂的线粒体呈碎片状或小而圆形,平均长度约2.83 μm。
2 线粒体的融合与分裂
线粒体动力学包括线粒体的融合与分裂,线粒体动力蛋白家族均含有三磷酸鸟苷(GTP)酶活性。介导线粒体融合的蛋白有线粒体融合蛋白(Mfn)1、Mfn2及视神经萎缩蛋白1(Opa1)(图1B)。线粒体分裂是一个多步骤的过程,包括线粒体膜的收缩及断裂,这一过程由Drp1及其受体介导,其受体包括线粒体分裂蛋白1(Fis1)、线粒体裂变因子(Mff)、49 kD线粒体动力蛋白(MiD49)和51 kD线粒体动力蛋白(MiD51)(图1C)。线粒体融合与分裂之间的动态平衡不但有利于线粒体的更新及清除,而且在维持心肌细胞收缩功能和能量代谢平衡中也发挥着重要作用[9]。
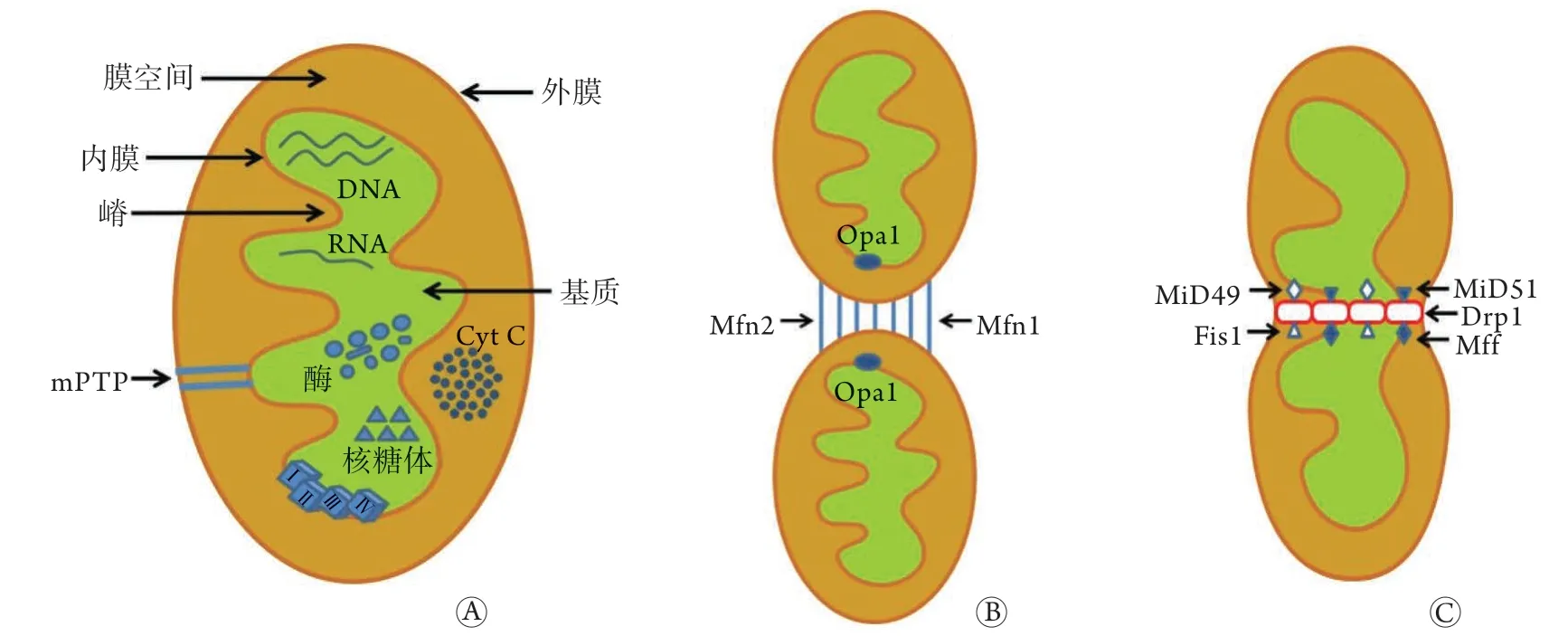
图1 线粒体的结构(A)、融合(B)与分裂(C)Fig.1 Mitochondrial structure(A), fusion (B) and fission (C)
3 Drp1与线粒体分裂
Drp1由4个保守区域组成,包括GTP酶结构域、中间结构域、可变结构域和GTP酶效应结构域(GED)。Drp1是一种胞质蛋白,通常以无活性的形式存在于细胞质中。Drp1活化后与Fis1、Mff结合,从胞质转运至线粒体外膜,在膜周围形成环状寡聚体结构,促进线粒体分裂。Drp1与线粒体外膜的结合受多种受体(如Fis1、Mff、MiD49、MiD51等)调节。MiD49及MiD51较Fis1、Mff招募Drp1的能力更强,二者可在无Fis1及Mff协助的情况下将Drp1招募至线粒体外膜,启动分裂。
Drp1的激活和转位受磷酸化、棕榈酰化、泛素化和亚硝酰化的调节,以磷酸化最为重要。Drp1的两个丝氨酸残基(ser616和ser637)通过磷酸化来影响Drp1的活性和定位。Drp1 ser616磷酸化后激活Drp1,使其向线粒体外膜转移,促进线粒体分裂;而Drp1 ser637磷酸化后则可抑制Drp1的活化及Drp1向线粒体外膜的转移,进而抑制线粒体分裂[10-11]。
高血糖、高血脂、缺血缺氧、紫外线辐射等均可使Drp1 ser616磷酸化,促进线粒体分裂。在2型糖尿病(T2DM)中,Drp1、Fis1、Mff的表达水平升高,而Mfn1、Mfn2、OPA1的表达水平明显降低,线粒体分裂增加,线粒体的形态表现为碎片化,且更短更圆。
4 Drp1与DCM
目前认为,氧化应激、代谢紊乱、心肌纤维化、炎症反应、细胞凋亡、内质网应激等是DCM细胞损伤的主要机制。最新研究发现,Drp1可参与糖尿病心肌细胞的活性氧(ROS)产生、能量代谢异常、糖脂代谢紊乱、胰岛素抵抗及细胞凋亡过程,导致DCM的发生发展,最终进展为心功能不全[12-13]。
4.1 Drp1与氧化应激 烟酰胺腺嘌呤二核苷酸(NADH)和黄素腺嘌呤二核苷酸(FADH2)氧化的电子通过线粒体内膜中的电子传输链(ETC)穿过内膜到达膜间隙,产生电化学梯度,促进无机磷酸与二磷酸腺苷(ADP)结合生成ATP。在糖尿病心肌细胞中,Drp1表达增高,线粒体复合体Ⅰ/Ⅱ/Ⅲ/Ⅳ的功能发生障碍,泛醌自由基延长,从而引起ETC异常电子泄露,并与氧原子结合后导致ROS产生增加[14]。同时,由于胰岛素缺乏或胰岛素抵抗,线粒体代谢底物从葡萄糖转换为脂肪酸,以代偿产生足够的ATP,但这个过程会产生更多的ROS,破坏氧化磷酸化过程[15]。
线粒体是氧化磷酸化的场所,也是ROS的主要来源,低浓度ROS具有细胞内信号转导功能,可发挥信号分子的作用。但高浓度的ROS可诱导细胞应激损伤甚至造成细胞死亡,导致DCM。Drp1过表达或Mfn1敲除后,线粒体分裂增加、融合减少,导致线粒体碎片化,并产生过量的ROS[16];ROS可增加解偶连蛋白(UCP)的活性,后者可使ATP生成和电子转运分离,导致热量产生增加而ATP生成减少[17]。其次,ROS还可损伤DNA、RNA、脂质、蛋白质及酶,导致糖尿病心肌细胞凋亡增加及DCM的发生。
线粒体不仅是ROS的主要产生部位,也是应激状态下ROS主要的攻击靶点[18]。Drp1过表达产生过量ROS,可导致氧化应激损伤、线粒体膜去极化增加、膜电位下降,并促进线粒体通透转换孔(mPTP)的病理性开放,膜间隙细胞色素C渗漏,进而出现线粒体肿胀、膜破裂及线粒体功能障碍,最终导致心肌细胞死亡[19-20]。氧化应激被认为是糖尿病心肌细胞损伤和DCM发生的罪魁祸首[21-23]。过量的ROS可耗尽细胞内的超氧化物歧化酶[锰超氧化物岐化酶(MnSOD)、铜锌超氧化物岐化酶(CuZnSOD)]和抗氧化酶[过氧化氢酶(CAT)、谷胱甘肽过氧化物酶(GPX)和过氧化物还原酶(PRX)],导致氧化应激损伤,损害心肌细胞Ca2+泵活性,同时也会降低心肌细胞对Ca2+的敏感性,抑制心肌收缩和舒张功能,从而导致心功能障碍及DCM进展[15,24]。
4.2 D r p 1 与能量代谢障碍 心肌细胞能量60%~80%来源于线粒体脂肪酸β-氧化。糖尿病初期,线粒体Drp1表达增高,线粒体分裂增加,葡萄糖氧化磷酸化受损,ATP产生减少,但由于RAAS激活,儿茶酚胺分泌增多,脂肪组织动员加速,血浆游离脂肪酸(FFA)浓度升高,FFA进入心肌细胞增加,脂肪酸β-氧化增强,代偿性产生足够的ATP以维持心肌细胞功能。但随着病情进展,Drp1表达进一步增高,线粒体分裂增加呈碎片状,线粒体复合体Ⅰ/Ⅱ/Ⅲ/Ⅳ的活性明显降低,呼吸链功能受损,线粒体功能障碍,脂肪酸β-氧化能力下降,ATP生成明显减少,导致糖尿病心功能障碍加重。此外,由于ATP合成减少,心肌细胞钙泵功能障碍,导致胞质Ca2+超载,进而损害心肌舒张及收缩功能,加重DCM心功能障碍[21,25-26]。
4.3 Drp1与细胞凋亡 与健康人相比,糖尿病心肌细胞凋亡增加了85倍,而线粒体功能障碍在糖尿病心肌细胞凋亡中发挥了重要作用[27-28]。DCM的特征是氧化应激和线粒体功能障碍,二者共同导致心肌细胞凋亡增加[29]。糖尿病线粒体Drp1 ser616磷酸化增加,活化的Drp1由胞质向线粒体转移,促进线粒体分裂,进而诱发线粒体内膜mPTP开放、膜电位下降、基质肿胀、线粒体外膜通透性增加,以及细胞色素C被释放至细胞质中,从而使凋亡因子caspase-3、B细胞淋巴瘤2相关X蛋白因子(Bax)活化,启动细胞凋亡程序;同时,ROS产生增加、氧化应激增强,线粒体呼吸链受损、ATP产生减少,内质网应激、炎症细胞因子释放增加,以及RAAS局部激活增加等因素,最终共同导致心肌细胞凋亡增加[30-33]。心肌细胞凋亡增加导致细胞数量减少,心肌收缩功能受损,最终造成心肌重塑、心脏扩大、心功能障碍,导致DCM恶化[34]。
4.4 Drp1与胰岛素抵抗和脂毒性 糖尿病心肌细胞中,Drp1、Fis1表达增多,Mfn1、Mfn2表达减少,线粒体分裂增加,碎片化线粒体增多,管状网络线粒体减少,线粒体动力学失衡。线粒体过度分裂可抑制胰岛素受体底物(IRS1)/蛋白激酶B(Akt)信号通路,并阻止葡萄糖转运体(GLUT)转移至细胞膜转运葡萄糖分子,从而导致胰岛素抵抗。同时,ROS产生增多也可抑制IRS1/Akt信号通路,导致GLUT转运葡萄糖减少,进一步加重胰岛素抵抗。由于胰岛素信号被抑制,GLUT向胞膜募集减少、葡萄糖摄入及氧化磷酸化减少,肌质网钙泵活性下降,心肌细胞内Ca2+浓度升高,导致心肌僵硬,最终发生DCM舒张功能不全[35]。
糖尿病心肌细胞中,由于胰岛素缺乏或胰岛素抵抗,心肌细胞葡萄糖摄取及利用率降低,心肌细胞摄取脂肪酸和β-氧化相对增加,以维持足够的ATP生成[36]。然而,心肌细胞摄取脂肪酸超过了β-氧化的能力,胞质内未完全代谢的脂肪酸增多,从而导致细胞内脂质蓄积和脂毒性增加。其中,二酰甘油(DAG)可通过激活蛋白激酶C(PKC)抑制胰岛素受体,神经酰胺(CER)也可通过抑制Akt而抑制胰岛素信号,诱发胰岛素抵抗。胰岛素抵抗和脂毒性共同导致心肌细胞能量代谢障碍,使心肌肥大、心腔扩大,DCM进一步加重[37-39]。
Drp1转运至线粒体是线粒体分裂的标志,也是发生胰岛素抵抗的必要条件。敲除Drp1或应用Drp1抑制剂阻止Drp1由胞质向线粒体外膜的转运可抑制线粒体分裂、减少线粒体碎片及延长线粒体网络,从而逆转胰岛素抵抗[40]。
5 Drp1抑制剂与DCM
线粒体分裂抑制剂1(Mdivi-1)可通过减少Drp1 ser616磷酸化来抑制Drp1活化,或通过抑制Drp1的GTP酶活性来抑制Drp1的功能,减少线粒体分裂,从而改善线粒体功能、抑制mPTP开放,减少心肌细胞凋亡。研究发现,Mdivi-1可抑制Drp1活化,使ROS和超氧阴离子产生减少,心肌细胞线粒体复合体Ⅰ/Ⅱ/Ⅲ/Ⅳ的功能改善,线粒体电子传输链ETC活性增强,ATP生成增多,DCM心功能改善[41]。
褪黑素通过SIRT1-PGC1α途径抑制Drp1的表达,并以腺苷酸活化蛋白激酶α(AMPKα)依赖的方式抑制Drp1 ser616的磷酸化,增强Drp1 ser637的磷酸化,从而抑制Drp1的活化、增加Mfn2和Opa1的表达,并可抑制线粒体分裂、增加线粒体融合,抑制mPTP开放、减少细胞色素C释放,减少线粒体膜电位下降及改善线粒体复合体Ⅰ/Ⅱ/Ⅲ/Ⅳ的功能,最终减少心肌细胞凋亡,改善DCM的心功能[42]。
黄芩苷是一种天然的黄酮化合物,可通过减少Drp1 ser616的磷酸化抑制Drp1活化,从而抑制线粒体分裂,并可减轻线粒体肿胀及膜电位下降,抑制心肌细胞凋亡,进而保护心功能,改善DCM的预后[30]。因此,Drp1抑制剂可能成为DCM的一种潜在治疗手段。
6 Drp1敲除的利与弊
线粒体自噬是清除细胞内异常线粒体的一种防御机制,通过线粒体不对称分裂选择性地清除老化、病理性及受损的线粒体,并通过溶酶体进行降解,是心肌细胞功能自我调节的关键[43-44]。Drp1介导的线粒体分裂与线粒体自噬之间存在相互依赖的关系。Drp1向线粒体募集既是启动分裂的标志,也是启动自噬的关键步骤[11,45]。DCM中,Drp1 Ser616磷酸化增加,Drp1转移至线粒体增多,同时线粒体自噬也被激活。但是,通过过度下调Drp1的表达来抑制Bnip3诱导的线粒体自噬,可导致细胞内受损线粒体清除减少,反而会加重线粒体功能障碍[46]。
糖尿病心肌细胞中Drp1表达增高,线粒体过度分裂,碎片化的线粒体成为线粒体自噬和溶酶体降解的目标,而有功能的线粒体相对减少,从而导致线粒体功能障碍。但是,过度抑制Drp1的表达也可能导致线粒体自噬被抑制,甚至造成心肌细胞损伤[47]。
研究发现,线粒体分裂是线粒体自噬的先决条件。敲除新生小鼠的Drp1可导致线粒体分裂减少、自噬受损,使受损线粒体蓄积、氧化应激增加、ATP生成减少、钙泵功能障碍,从而使细胞内脂质过氧化物含量升高,细胞电活动受损,以及白细胞介素(IL)-6、肿瘤坏死因子(TNF)-α等炎性因子分泌增加,进而导致严重的肌肉萎缩,危及新生小鼠的心脏发育,最终发生心腔扩张和心力衰竭[48]。
7 总结与展望
糖尿病心肌细胞中Drp1活化增强,线粒体分裂增加、融合减少,线粒体功能障碍,导致心肌细胞氧化应激增强、能量代谢障碍、细胞凋亡增加、胰岛素抵抗和脂毒性增加,进而诱发糖尿病心肌病的发生发展。抑制Drp1的活化及表达,可减少心肌细胞凋亡,改善DCM心功能,以及延缓病情进展。然而,敲除Drp1后线粒体分裂减少、自噬受损,线粒体功能障碍加重,则可导致心肌细胞受损、心功能障碍加重。因此,线粒体分裂是一把双刃剑,生理性分裂对于线粒体功能至关重要,抑制Drp1的过度活化,保持线粒体分裂与融合之间的动态平衡,才能维持正常的线粒体功能及糖尿病和DCM患者的心脏功能,并改善疾病预后。但是,如何通过Drp1精准平衡线粒体分裂与融合过程,仍需要更多的探索性研究进行验证。
猜你喜欢
材料与冶金学报(2022年2期)2022-08-10
当代陕西(2022年5期)2022-04-19
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
科学与财富(2021年33期)2021-05-10
现代装饰(2021年1期)2021-03-29
青岛大学学报(医学版)(2021年1期)2021-03-03
心肺血管病杂志(2020年5期)2021-01-14
体育科学(2018年12期)2019-01-04
分析化学(2017年12期)2017-12-25
