
基于多重解码器的自编码器模型的生物序列聚类方法
2022-11-15 14:11陈城,林劼
福建师范大学学报(自然科学版) 2022年6期
陈 城,林 劼
(福建师范大学数学与统计学院,福建 福州 350117)
生物测序技术的发展产生大量的生物序列数据,对序列进行聚类分析,把功能相近的序列聚为一类,可以帮助科研人员快速了解生物序列的功能,为后续的研究奠定坚实的基础.聚类在生物信息学中并不是一个新话题.基因表达数据的分析常对微阵列数据进行聚类,并认为在同一个簇中的基因具有相同的功能.学者们提出了几种先进的微阵列聚类算法[1],包括层次聚类[2]和集成聚类[3].为了避免微阵列数据中的噪声,双聚类[4]的方法应运而生,它能够同时进行特征选择和样本选择.单细胞测序数据的工作原理类似于基因表达数据,根据不同的细胞类型[5-6]对不同的基因表达进行聚类.然而,这些方法关注的是基因表达数据,而不是生物序列数据.
生物序列聚类是聚类算法在生物学中的应用,使用的数据为:核酸序列(如DNA、RNA序列)或氨基酸序列(如蛋白质序列).其中,核酸序列由4种碱基组成:A(腺嘌呤)、G(鸟嘌呤)、C(胞嘧啶)、T(胸腺嘧啶)/U(尿嘧啶),蛋白质序列则是由20种氨基酸排列组合而成.序列聚类需要将序列转化为数值向量.虽然有一些工具可以将DNA/RNA/蛋白质序列转化为数值向量[7-10],但对序列进行表示学习仍然是聚类过程中重要的一步.生物序列聚类研究构建衡量序列间的相似性或差异性的数学模型,旨在根据统计结果将序列划分为几个簇,同一簇中的序列具有相似的功能,这样可以由已知序列的功能推测未知序列的功能[11].
生物序列聚类有利于分析序列潜在的结构、功能等信息,进一步推演序列在进化过程中发生的先后关系[12].迄今为止,学者们已经提出众多优秀的聚类算法,但是由于领域的差异、解释性差等问题,不能直接应用在生物序列聚类上.目前,生物序列聚类的困难之处在于生物序列数据的特征难以正确提取,数据量大,计算复杂度高,内存需求高,不能保证一定能够找到最优解且很难从生物学角度进行结果解释等.
本文提出一种基于多重解码器的自编码器模型,用于学习生物序列数据的表示,然后使用 k-means算法对序列的表示进行聚类. 试验结果验证本文提出的方法在 DNA 序列数据集上具有良好的性能.
1 相关工作
根据聚类方法的不同,生物序列上的聚类研究大致可分为:启发式生物聚类算法、层次生物聚类算法和其他生物聚类算法.
1.1 启发式生物聚类算法
基于启发式的生物聚类算法采用一种简单贪婪策略,始于一个种子(seed),基于一定的搜索技术,合并、扩张,完成序列的聚类过程.其代表性算法为FastGroup[13]、CD-HIT[14]、UCLUST[15]等.
2001年,Seguritan首次提出一种基于启发式的生物序列聚类算法FastGroup.该算法主要有3个步骤:首先将数据集中的所有序列相互比较;然后将相似的序列分组;最后从每组中输出一个代表序列.CD-HIT使用贪婪的增量聚类算法,该算法通过短词过滤策略和并行化技术得到提高,有效地对大规模序列数据集进行聚类.短词过滤策略的整体思想为:若序列与代表序列的相似性低于预先设置的阈值,则该序列不进行序列比对.该策略可以使算法的运行速度加快,其复杂度为O(N),N为序列数量.
USEARCH算法在进行比对时会在所有相似度达到阈值的序列中寻找合适的比对位点,基于这个特点,UCLUST提出一种改进方法,仅需要寻找一个或几个合适的比对位点,减少了比对位点的搜索数量,提高了序列比对速度.与CD-HIT相比,这种方法的运算速度快、内存需求低,且灵敏度较高.
Ghodsi等[16]提出了新的词过滤方法DNACLUST,避免了序列间的两两比对,是一种贪婪算法.该算法在精确模型下的运算速度优于CD-HIT与UCLUST,但在近似模型下的运算速度与UCLUST差不多.SEED[17]则使用开放哈希技术和一种特殊类型的称为块间隔的种子将输入序列聚类.
UPARSE[18]是来自USEARCH的最新从头聚类方法,它通过质量过滤算法过滤read,将其修整为固定长度.该方法使用UPARSE-OTU进行聚类,这是一种新的贪婪算法,可同时执行嵌合过滤和OTU聚类.由于质量过滤算法的严格,生物物种丰富度和多样性会被显著低估,最后产生的生物学结果存在较多错误.大规模过滤序列能够减少运行时间,但是过滤参数需要针对数据的不同进行改变,且不能自动对过滤参数进行选择,需要多次试验后人为进行选择,而这个过程的时间成本可能很高.
在进行聚类的过程中,启发式算法不需要计算距离矩阵,因此降低了存储空间的需求和计算复杂度.缺点是不能保证一定能够找到最优解.
1.2 层次生物聚类算法
基于层次的生物序列聚类算法通过序列比对,根据一定的相似性或距离度量方式获得距离矩阵,再采用贪婪层次聚类算法完成生物序列聚类,是目前最常用的生物序列聚类方法,其代表性算法为DOTUR[19]、Mothur[20]、ESPRIT[21]、mBKM[22]等.
2005年,Schloss提出一种无种子(seed)的生物序列聚类算法DOTUR.DOTUR使用序列之间的遗传距离将序列分配给OTU.它通过使用最远、平均或最近邻居算法为OTU分配序列,并估计一个簇的丰富性和多样性.
在DOTUR的基础上,Schloss又开发了Mothur.Mothur被用于修剪、筛选和排列序列,计算距离,将序列分配给OTU,Alpha和Beta多样性计算,序列比对,序列聚类注释.距离矩阵对于Mothur聚集OTUs很重要.矩阵可以反映每个序列与其他序列的相似性或距离.计算矩阵和聚类具有较高的时间复杂度.基于此,Mothur不适合处理大数据集[23].该算法存在假阳性率高、噪音信号强等缺点,且很难从生物学角度进行结果解释.
Sun采用ESPRIT算法对生物序列进行聚类划分.该算法由4个模块组成:(1)使用各种标准去除低质量read;(2)计算read的成对距离;(3)将read分组到不同差异水平的OTU中;(4)执行统计推断来估计物种丰富度.该算法使用了Needleman-Wunsch双序列比对算法,以k-mers的形式过滤无需比对的序列,利用在线学习开发了一种名为Hduster的方法进行层次聚类,在一定程度上降低了计算机的内存需求,但计算复杂度为O(N2),当N较大时,算法的时间成本高.
Cai[24]基于ESPRIT算法提出了一种新的在线学习的算法ESPRIT-Tree.基本思想是使用伪度量构造分区树,利用分区树将序列空间划分为一组子空间,然后递归地细化这些子空间中的簇结构.该技术依赖于快速最近对搜索和一种动态插入和删除树结点的方法.为了避免穷举计算簇之间的成对距离,该方法将序列的每个簇表示为概率序列,并进行一系列操作来比对这些概率序列并计算它们之间的遗传距离.ESPRIT-Tree是启发式生物序列聚类算法的一种,该算法同时解决了计算复杂度高和计算机内存需求多的问题,其计算复杂度几乎与生物序列数目呈线性关系.
mBKM是一种基于新的距离度量DMk的非比对算法,用于聚类基因序列.该方法将DNA序列转化为特征向量,其中包含DNA序列中k-mer的出现次数、位置和顺序关系.然后,将层次聚类算法应用于DNA序列.研究表明[25-26],当基于同质性标准对数据集进行划分时,该方法得到了较好的聚类结果.
层次生物序列聚类算法的缺点是在拥有庞大的序列数据时,所需要的储存空间大,计算复杂度高.
1.3 其他生物聚类算法
其他生物聚类算法,如CROP[27]算法利用等级机制将需要比对的序列划分为若干个子集,然后基于贝叶斯理论,采用高斯混合模型对序列进行聚类.在生物序列中,CROP算法可以推断出最优的聚类结果,其中高斯模型的抗噪声能力能够克服由于测序误差而导致对序列的高估、内存需求高及计算效率低的问题,在一定程度上能够较好地实现生物序列聚类,具有较强的抗噪声能力和鲁棒性.
CBE[28]是一种基于最大熵原理的聚类方法.这种方法基于数据的先验信息来探索数据所有可能的概率分布空间,得到熵最大的分布,当熵值达到最大时,聚类结束.先验信息基于以下假设:根据某种统计方法,簇中元素彼此相似.基于此,满足条件的那些高熵分布优于其他分布.
coreClust[29]是一种基于检测保守区域的非比对聚类方法.这些区域的检测可用于功能注释和分组蛋白质序列的区域.coreClust基于一种名为MinHash的技术,该技术是一种局部敏感哈希方法,用于识别集合中的相似元素.它主要依赖于哈希,因此该方法非常适合MapReduce并行处理平台,从而实现可伸缩性.

DeLUCS[33]模型使用DNA序列的频率混沌博弈表示(frequency chaos game representations,FCGR),并通过优化多个神经网络生成模拟序列的FCGRs来学习数据的模式(基因组签名).然后使用多数投票方案来确定每个序列的最终簇分配.ALFATClust[34]利用快速成对非比对的序列距离计算和社区检测来生成簇.ALFATClust可以通过考虑簇的分离和簇内序列相似性来动态确定生成每个簇的阈值,而不是对每个生成的簇应用单个阈值.
2 方法
本文提出一种基于多重解码器的自编码器模型,用于生物序列数据的表示学习,然后使用k-means算法对序列的表示进行聚类.将整个过程分为两个阶段:表示学习阶段和聚类阶段.
2.1 表示学习
在进行聚类之前,需要对生物序列进行表示学习,得到序列所对应的表示,以便进行后续的聚类任务.本方法使用的表示学习模型如图1所示.首先,对生物序列数据进行k-mer划分并统计k-mer的频率,得到序列k-mer的频率向量作为模型的输入;其次,在自编码器模型的基础上应用多重解码器的形式,从多个不同的空间对特征进行重构;最后,将序列的k-mer频率向量输入到模型中使用均方差损失进行训练得到序列的表示.

图1 基于多重解码器的自编码器模型
2.1.1 k-mer的提取和频率向量
k-mer是一段长度为k的子串,它是由序列剪切一部分得到的.k-mer的提取指的是将一条序列连续切割,按照碱基的排列顺序依次划动得到的一系列长度为k的子串.
给定生物序列集S={S1,S2,…,Si,…,SN},序列Si的长度为M,在序列上通过一个长度为k(2≤k≤M-1)、步长为1的滑动窗口从序列中提取长度为k的子串,即k-mer.在长度为M的序列中存在M-k+1个k-mer.一条具有|Σ|个字母的序列最多有|Σ|k种不同的k-mer.一条DNA序列由字母表中的字母组成,字母表Σ={A,C,G,T}且|Σ|=4.
使用单个字母的频率作为特征表示样本时,所有的关联信息会被丢失.k-mer频率向量通过描绘序列局部信息将每条样本转化成|Σ|k维向量,从而弥补这一缺陷.因此,使用k-mer频率向量来表示样本序列.
具有|Σ|k维的k-mer频率向量可以表示为:
Xi= [Xi1,Xi2,…,Xij,…,Xi|Σ|k]T,
(1)
其中,Xij是第i条序列中第j种k-mer出现的频率,Xij可由公式(2)计算得到:
(2)
其中,Nij是在第i条序列提取的所有的k-mer中第j种k-mer出现的次数,Ni是第i条序列中提取的所有k-mer的数量.
2.1.2 编码器
对生物序列Si提取k-mer得到频率向量Xi后,使用编码器E(·)对k-mer频率向量Xi进行特征提取,如公式(3)所示:
Zi=E(Xi),i=1,2,…,N,
(3)
其中,Zi表示序列Si的特征向量.在本文中,使用编码器E(·)对序列数据进行映射,得到序列的表示.它需要与解码器Dl(·)一起使用均方差损失进行训练.训练完成后,不再使用解码器的部分,而是单独使用编码器.由编码器得到的表示可以用于下游任务,本文中,应用学习到的表示进行聚类.
2.1.3 多重解码器
与传统的自编码器模型不同,本文采用多重解码器的形式,具有提高编码器表征能力的优点.给定L个解码器Dl(·),l=1,2,…,L,对于序列Si的特征向量Zi,使用解码器对其进行解码,重构序列Si的k-mer频率向量Xi,如公式(4)所示:
(4)

2.1.4 学习策略
综上所述,模型是由一个编码器和多个解码器组成.训练时,分别使用多个解码器的输出和序列的k-mer频率向量计算均方差损失,解码器Dl(·)的损失函数Ll如公式(5)所示:
(5)

最后,同时最小化解码器的均方差损失,总损失函数如公式(6)所示:
(6)
模型训练完毕后,使用编码器的输出Zi作为序列的表示进行后续的任务.
2.2 聚类
为了推测未知生物序列的功能,分析基因间进化的先后顺序关系,对生物序列进行聚类有助于科研人员快速了解生物序列,加快推动相应研究的进展.本文使用的聚类算法是k-means算法.该算法具有收敛速度快的优点,并且能够达到较好的聚类效果.
k-means算法是最常用的聚类算法,主要思想是在给定k值和k个初始簇中心点的情况下,把每个点(即样本)分到离其最近的簇中心点所代表的簇中,所有点分配完毕后,根据一个簇内的所有点重新计算该簇的中心点(取平均值),然后再迭代进行分配点和更新簇中心点的步骤,直至簇中心点的变化很小,或者达到指定的迭代次数.
给定样本的表示Z(即为编码器的输出),包含了n个样本的表示Z={Z1,Z2,Z3,…,Zn},其中每个表示都具有m个维度的特征.对于k-means算法,首先需要初始化k个簇中心{C1,C2,C3,…,Ck},1 (7) 其中,Zi表示第i个对象,1≤i≤n,Cj表示第j个簇中心,1≤j≤k,Zit表示第i个对象的第t个分量,1≤t≤m,Cjt表示第j个簇中心的第t个分量. 依次比较每一个样本到每一个簇中心的距离,将对象分配到距离最近的簇中心的簇中,得到k个簇{S1,S2,S3,…,Sk}. 聚类时,目标是最小化每个样本到其对应的簇中心的距离,如公式(8)所示: (8) 基于多重解码器的自编码器模型的生物序列聚类算法,如算法1所示. 算法1 基于多重解码器的自编码器模型的生物序列聚类算法输入:生物序列集S,编码器E(·),解码器Dl,序列数量N,解码器数量L输出:生物序列的簇标签1:for i=1 to Ndo2: Xi=从Si中提取kmer的频率向量3: Zi = E(Xi)4: for l=1 to L do5: ^Xli=Dl(Zi)6: end for7:end for8: 计算损失 Ltotal如公式(6)所示9: 使用随机梯度下降法计算梯度10:更新模型参数,直到收敛11:得到训练完毕的编码器 E(·)12:for i=1 to N do13: Zi= E(Xi)14:end for15: 使用k-means 对序列表示集 Z进行聚类,得到生物序列的簇标签 HOGENOM是一个全序列生物同源基因家族的数据库,其中包含来自真核生物、细菌和古细菌的182个完整基因组的同源基因家族.本选题使用从HOGENOM中随机抽取的HOG100、HOG200、HOG300共3个DNA序列数据集,其对应的序列类别数分别为100、200和300,这些DNA数据由蛋白质生物学和化学研究所(IBCP)使用基于人口的增量学习(PBIL)进行收集,可以从ftp:∥pbil.univlyon1.fr/pub/hogenom/release—06/获得相关数据.表1列出了这3个DNA数据集的详细信息. 表1 DNA数据集的详细信息 本文选择的k-mer的k为3.编码器的网络结构为3个全连接层,神经元参数分别设置为128、64和16,激活函数均为relu函数.解码器D1(·)的网络结构为4个全连接层,神经元参数分别设置为128、64、32和64,激活函数均为relu函数;解码器D2(·)的网络结构为5个全连接层,神经元参数分别设置为128、64、56、32和64,激活函数均为relu函数;解码器D3(·)的网络结构为5个全连接层,神经元参数分别设置为128、256、128、32和64,第一层和最后一层的激活函数为tanh函数,其余均为relu函数. 本文采用的评价指标是归一化互信息 (normalized mutual information,NMI),NMI是聚类评价指标中的一种外部指标,用于度量聚类结果与真实标签之间的关系,其范围为[0,1].当聚类的结果越接近真实的情况,NMI的数值就越接近1.若两者之间相互独立,那么NMI就为0.NMI的定义如公式(9)所示: (9) 其中,I表示互信息(mutual information),H为熵. (10) 其中,P(wk)、P(cj)、P(wk∩cj)可以分别看作样本属于聚类簇wk,属于类别cj,同时属于两者的概率.第二个等价式子则是由概率的极大似然估计推导而来. (11) 表2 聚类结果的比较 从试验的结果可以看出,本文提出的方法在HOG100数据集上具有竞争性,在其他2个数据集上性能优于另外2个方法.随着数据集类别数量的增加,本方法的表现更加稳定,其他2种方法的性能反而出现下滑的趋势,说明本方法在种类数量大的序列数据集中一样适用. 本文提出了一种基于多重解码器的自编码器模型的聚类方法.在自编码器的基础上进行了改造,增加了解码器的数量,从不同的空间对输入数据进行了重构,从而加强了模型进行表示学习的能力;接着使用模型中编码器的输出作为k-means的输入来进行聚类.试验表明本方法获得了良好的聚类效果,而且在当前数据集下随着数据集的类别数量的增加,本方法的性能将趋于稳定. 未来会进一步针对解码器研究适用于不同任务的学习策略,继续提高编码器的表征能力,使得学习到的嵌入根据学习策略的不同能够满足一个或多个下游任务的需求.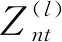

3 数据试验
3.1 数据集

3.2 参数设置
3.3 评价指标
3.4 试验结果

4 结论
猜你喜欢
网络安全与数据管理(2022年1期)2022-08-29
小学生必读(低年级版)(2021年10期)2022-01-18
小学生必读(低年级版)(2021年11期)2021-03-09
小学生必读(低年级版)(2021年12期)2021-03-04
家庭影院技术(2019年8期)2019-12-04
铁道通信信号(2019年6期)2019-10-08
制造技术与机床(2017年7期)2018-01-19
雷达学报(2017年6期)2017-03-26
互联网天地(2016年1期)2016-05-04
智能系统学报(2015年4期)2015-12-27
