
HEXA 基因变异GM2婴儿型神经节苷脂沉积病1例诊疗分析
2022-11-26 10:23吴双凤黄琳贺兵
淮海医药 2022年5期
吴双凤,黄琳,贺兵
神经节苷脂存在于人体的各种细胞,而以脑组织含量最高。神经节苷脂沉积症临床分多种亚型,其中以GM1、GM2节苷脂沉积症较常见。GM2节苷脂沉积病为常染色体隐性遗传,由己糖胺酶缺乏所致。该酶有两种同工酶,分别是己糖胺酶A(HexA)和己糖胺酶B(HexB)。B型GM2神经节苷脂沉积症(Tay-Sachs病)是HEXA基因的遗传变异导致β-己糖胺酶A(β-hexosaminidase A,HEXA )同工酶活性缺乏,从而引起GM2在神经元积累,以脑和脊髓神经细胞进行性破坏为特征,是目前最早被发现的严重溶酶体疾病之一[1]。婴儿型的Tay-Sachs病初生时均正常,4月左右出现对声音刺激特别敏感,至4~6月时呈现精神运动发育方面的衰退征象,至8~9月时,患儿可出现失明,生后第二年常有癫痫发作和脑电图异常表现,随着病情的进展患儿渐呈痴呆状,常在3~5岁死亡。
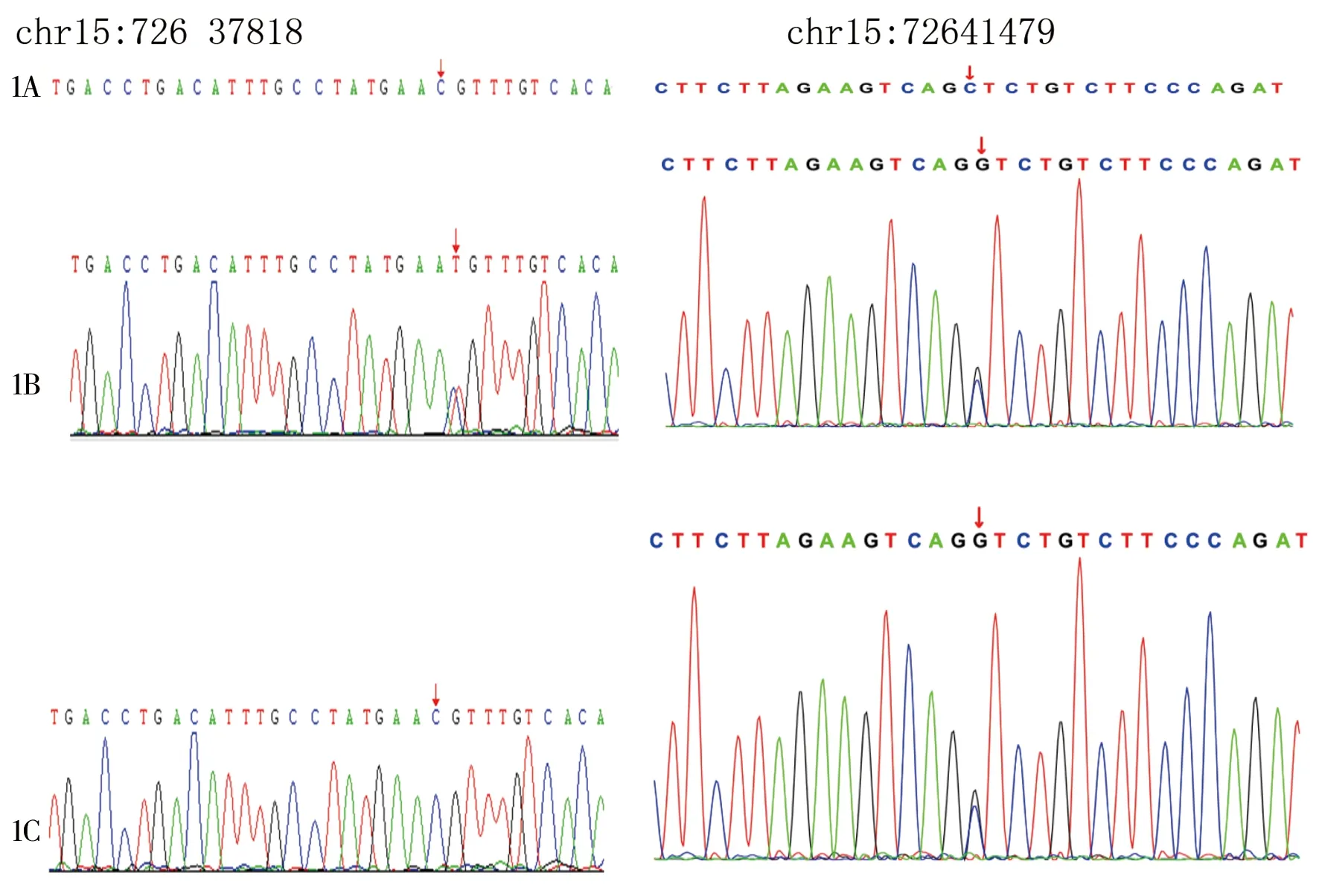
1A HEXA:c.1495(exon13)C>T及 HEXA:c.927(exon8)C>G的标准序列;1B 患儿chr15:72637818存在c.1495(exo n13)C>T 的杂合突变,chr15:72641479存在c.927(exon 8)C>G杂合突变;1C 患儿之母chr15:726 37818无突变,chr15:72641479存在c.927(exon8)C>G杂合突变;图1 患儿及父母HEXA基因c.1445A>T和 c.1052T>C突变位点Sanger测序结果
1 病例资料
1.1 临床资料 患儿,女,1岁10月,因精神运动发育倒退1年,反复哭闹3天,间断抽搐3小时于2021年4月20日在湖南邵阳学院附属第二医院就诊,患儿系为G2P2足月剖宫产出生,出生时无窒息病史,出生时体质量3.2 kg,出生10月内患儿身高、体质量基本接近正常儿童,2月会笑,3月抬头,4~6月应人反应正常,6月会坐,8月会爬,但患儿一直有阵发性肢体抖动现象,患儿10月时出现运动、精神发育倒退,渐不能独坐及抬头,不能抓握,不能成单字说话及逗笑,目光逐渐呆滞,且喂奶时易呛咳,喂养困难。1岁时就诊当地卫生院,考虑“脑瘫”,一直未予重视,康复效果欠佳,入院前3天患儿出现反复哭闹,喂养困难,大便干结,阵发性喉中鸣响,自行予以“开塞露”通便,患儿无好转,入院前3小时患儿出现间歇性抽搐、全身呈角弓反张状,喉鸣加重,神智不转清,全身肌肉间歇性痉挛强直。患儿入院体查:T38.2 ℃,持续抽搐状态,口唇无发绀,牙关紧闭,口吐白沫,头后仰,呼吸急促,可见吸气性三凹征,双肺可闻及较多痰鸣音,双上肢肌张力增高,屈曲状,被动活动有折刀样抵抗感,双下肢肌张力明显增高,呈剪刀状,全身呈角弓反张状,病理征未引出。
1.2 实验室检查 (1)血常规(2021年4月20日):WBC:23×109/L,N:87%,HB:120 g/L,Plt:543×109/L,经抗感染治疗5天后恢复正常;(2)脑脊液常规、生化、压力均无异常。(3)肝功能、电解质、血糖、血脂、铜蓝蛋白、电解质及CRP、降钙素原均正常;心肌酶谱:CK:687.8 U/L,CK-MB67.97 U/L,LDH:1070 U/L,血清肌钙蛋白Ⅰ测定(cTnⅠ):7.61 ng/mL。(4)乳酸:2.3 mmol/L,(5)血液酶学分析示己糖胺酶A活力低,为0.2 nmol/(mg·h) [参考值为2.8~9.5 nmol/(mg·h)]。
1.3 影像学检查 (1)头颅MRI正常。(2)24小时脑电图:基本节律欠佳:双侧各区可见稍多慢波,对称欠佳,以右侧为甚,额、颞区为著,偶可见棘波。(3)心脏彩超:卵圆孔未闭声像,三尖瓣反流声像。
1.4 基因检测分析结果 (1)线粒体DNA高敏感性测序分析:未发现线粒体DNA致病性较高的变异位点。(2)取患儿EDTA抗凝外周血行加深全外显子组检测分析:结果显示发现HEXA 基因的2个变异,位于染色体位置chr15:72637818的核酸c.1495(exo n13)C>T 以及位于染色体位置chr15:72641479的核酸c.927(exon 8)C>G ,从而引起p.R499C(p.Arg499Cys)(NM_000520)氨基酸和p.S309R(p.Ser309Arg) (NM_000520) 氨基酸改变,其中c.927(exon 8)C>G来自于母亲(见图1)。根据ACMG指南(2015年),c.1495(exo n13)C>T变异可能致病,c.927(exon 8)C>G 变异不确定。很遗憾患儿家属因家庭原因(其父缺失),本次检测缺少家系成员(尤其是其父)携带信息,故无法判断遗传共分离。
1.5 治疗及转归 结合临床表现、酶学检查及基因分析该患儿确诊为Tay-Sachs病,入院后予以抗感染、丙戊酸、左卡尼汀治疗,并补充B族维生素,辅酶Q10治疗,患儿确诊后感染控制出院,出院带丙戊酸钠(2.5毫升/次,每天3次)口服,2021年10月25日微信追踪患儿目前仍有间歇惊厥发作,四肢呈折刀样,病情无明显好转。
2 讨论
GM2神经节苷脂病是溶酶体贮存障碍的一个亚群,由β-己糖胺酶的α和β肽链亚基因突变引起,包括Tay-Sachs病和Sandhoff病[1]。这种疾病的特点是GM2神经节苷脂在细胞内过度积累,从而引起一系列的临床症状。有研究[2]报道该病引起急性神经退变先于激活的小胶质细胞扩张、巨噬细胞和星形胶质细胞活化,同时伴随炎症递质的产生,也有研究[3]提示GM2的积聚会导致管腔内Ca2+的耗竭,进而激活三种UPR传感器之一的PERK信号通路,促凋亡转录因子C / EBP同源蛋白( CHOP )的表达增强,神经细胞内GM2的积累诱导神经突起萎缩和凋亡。多数文献报道Tay-Sachs病( TSD )是由编码HexosaminidaseA ( HEXA )酶的HEXA基因突变引起的,而HEXA基因位于15q23.3,包含14 个外显子,其突变可造成溶酶体β氨基己糖苷酶A酶缺陷[4-5],从而引起疾病发生。
根据发病时间,Tay-Sachs病通常被分为婴儿、青少年和成人的形式[1]。婴儿型Tay-Sachs病最为常见,常于婴儿期起病,在出生时表现起来很健康,但进行性GM2的积累会导致运动功能和认知能力的丧失、发育回归、肌张力障碍、失明、癫痫发作等[2]症状。主要表现为盲、痴呆、冷漠、苍白、癫痫发作、肌张力高、全身性低张力、对惊吓反应过度、精神运动功能衰退、头部控制差、误吸等[6-8]。我院收治该病例起病年龄较小,婴儿期一直有惊跳现象,但家属未予重视,后10月起发现精神运动、语言发育倒退,但被误诊为“脑瘫”。患儿家庭不和睦,经济情况落后导致患儿就诊时间较晚,就诊时神经发育进行性倒退现象引起接诊医生的注意,从而行全外显子组测序分析,方确诊。该患儿确诊时已有持续性惊厥发作、吞咽及喂养困难等严重表现,以神经系统受累为主,这和相关报道的“GM1神经节苷脂沉积病既有中枢神经系统表现,也可有全身表现,而GM2神经节苷病主要表现为中枢神经系症状”[9]相一致。但检验结果与其他文献[4,10]报道的Tay-Sachs病有所不同的是,该患儿的乳酸一直增高,可能和患儿一直处于惊厥状态有关。
Tay-Sachs病为常染色体隐形遗传病,患儿基因测序提示HEXA基因的两个位点均为杂合突变,位于染色体位置chr15:72637818的核酸c.1495(exo n13)C>T 为可能致病基因,其中有一个不确定致病基因chr15:72641479的c.927(exon8)C>G杂合突变可能来自于母亲。但其母携带不止一个杂合变异,不排除呈复合杂合携带(支持AR遗传病发病机制)的可能。因本次检测缺少家系成员(尤其是其父)携带信息,故无法判断遗传共分离。患儿临床表型与Tay-Sachs病匹配,早期有阵发性肢体抖动、目光呆滞,初期有斜视,但基层医生及家庭成员均未重视该病情,有研究报道眼底检查黄斑中樱桃红点的存在有助于对Tay-Sachs病患儿的早期诊断[8-9],对有相应神经症状的患儿,医生应考虑遗传代谢性疾病可能,早期进行眼底筛查及相关底物筛查。
GM2 神经节苷脂病是一个罕见病,其发病率常与种族人群相关,其中德系犹太人、法裔加拿大人、爱尔兰人、宾夕法尼亚荷兰人、印第安人等均为高危人群[11],德系犹太人群体已开展了孕前筛查[12]。目前国内关于Tay-Sachs病的临床病例报道并不多见,近5年报道了2例,分别是2021年7月吴沪军等[13]报道的1岁3月龄的患儿和杨志刚等报道的1岁7月龄患,均是复合杂合突变,其中杨志刚等[4]报道的病例在诊断3月后因呼吸困难家中死亡。本文患儿的 c.1495(exo n13)C>T 和c.927(exon8)C>G突变位点丰富了HEXA的基因突变谱,患儿的结局正在追踪中。对于该疾病的预防、逆转都尚处于研究阶段,酶置换治疗与细胞移植、底物还原、酶增强疗法都还没有明确的临床数据[14],利用病毒为载体传递编码α和βHexA亚基基因的治疗有可能在未来作为干预措施[9,15]。
猜你喜欢
中国临床医学影像杂志(2022年5期)2022-07-26
种子(2021年3期)2021-04-12
中国生殖健康(2020年4期)2021-01-18
中国现代中药(2019年5期)2019-07-03
科海故事博览·下旬刊(2019年6期)2019-04-16
中医眼耳鼻喉杂志(2019年2期)2019-04-13
中国生育健康杂志(2018年6期)2018-11-13
中国生殖健康(2018年4期)2018-11-06
科技视界(2016年27期)2017-03-14
中学生理科应试(2016年7期)2016-05-14
