
不同形貌Co3O4的制备及其活化过一硫酸盐降解亚甲基蓝的性能
2022-12-06 06:29廖小刚沈海丽
无机化学学报 2022年11期
夏 强 廖小刚 沈海丽 郑 林 李 纲*, 田 甜,2
(1重庆理工大学化学化工学院,重庆 400054)
(2中国兵器工业集团第五二研究所,包头 014000)
0 引 言
近来,基于过一硫酸盐(PMS)的高级氧化技术用于处理有机废水受到关注。该技术原理为通过一定的活化手段使PMS分解产生具有极强氧化活性的·SO4-等中间体,进而实现对废水中有机污染物的快速降解。在众多PMS活化手段中,催化活化因其活化效果显著、操作简单易行且无需能量消耗等优势,成为了目前研究的热点。过渡金属元素因具有可变的化学价态而表现出良好的催化活性,而其中Co被认为是催化活化PMS性能最为优异的元素。
PMS催化活化方式包括均相催化和非均相催化。与均相催化剂Co2+相比,非均相催化剂Co3O4因可从降解液中回收而更具实用化前景[1-4]。如Anipsitakis等[5]的研究表明,Co3O4作为催化剂使用时不仅表现出理想的PMS活化效果,而且在水溶液中的Co2+流失量小,对环境更加友好。Chen等[6]通过沉淀法制备的纳米Co3O4作为非均相催化剂具有优异的活化PMS降解酸性橙的性能,且在循环使用中稳定性保持良好。但非均相催化剂Co3O4用于活化PMS处理难降解有机污染物尚存在如下不足:一是与均相Co2+活化PMS的性能相比还存在较大差距;二是为了取得理想的有机污染物降解效果,投加至反应体系中催化剂的用量通常较大。这些Co3O4/PMS高级氧化(AOP)体系中存在的催化剂用量大、反应时间过长等问题,制约了其走向实际应用,而进一步提高催化剂的活性则是解决上述问题的关键。
通过构筑复合体系(如与生物炭等非金属[7-10]或MnO2等金属氧化物复合[11-13])是现有报道中用于改善Co3O4催化剂对PMS活化性能最常用的方法。然而,从材料制备方法及材料形貌本身出发来研究Co3O4与其活化PMS性能关系的报道则较为少见[14]。基于此,我们通过热解不同的含钴前驱体材料制备出3种不同形貌的Co3O4催化剂并将其用于对PMS的活化,借助多种表征测试手段深入分析了不同制备方法所获Co3O4的催化性能存在显著差异的原因。在此基础上,对筛选出的具有最优异活化PMS性能的Co3O4材料催化降解模拟染料废水(亚甲基蓝,MB)的效果进行考察。结果显示,采用化学浴沉积-煅烧法所获Co3O4材料具有的高活性使其在反应体系中的用量(质量浓度)大大减少至0.04 g·L-1以下,反应时间缩短为25 min,极大地提高了对有机废水的处理效果,为前述问题的解决提供了可供选择的方案。
1 实验部分
1.1 试剂与仪器
实验所用的试剂药品包括MB、过一硫酸氢钾、亚硝酸钠、尿素、七水合硫酸钴、六水合硝酸钴、浓氨水(25%~28%)、二水合草酸、乙二醇、碘化钾、碳酸氢钠、无水乙醇(EtOH)和叔丁醇(TBA),均为分析纯。实验用水均为去离子水,由GWA-UN2型超纯水器(北京普析通用仪器有限公司)制得。
样品的晶相结构和相纯度测试在XRD-7000型X射线衍射仪(XRD)上完成,Cu靶Kα辐射,X射线波长为0.154 06 nm,加速电压和电流分别为40 kV和30 mA,扫描范围2θ=10°~80°,扫描速率2(°)·min-1,扫描步长0.02°。样品的形貌通过日立Regulus8100型扫描电子显微镜(SEM)进行观察,加速电压为5 kV。样品的N2吸附-脱附曲线在Micrometritics ASAP 2020氮气吸附仪上完成,工作温度为-195.8℃。样品的X射线光电子能谱(XPS)测试在Kratos XSAM-800型X射线光电子能谱仪上完成,Al靶Kα射线作为发射源。样品氧空位检测和自由基捕获实验在JES FA200型电子自旋(顺磁)共振波谱仪(ESR/EPR)上完成,并选用5,5-二甲基-1-吡咯啉-N-氧化物(DMPO)作为·SO4-、·OH或·O2-捕获剂,4-氧-2,2,6,6-四甲基哌啶(TEMP)为1O2捕获剂。
1.2 实验步骤
1.2.1 催化剂的制备
Co3O4-A采用尿素水热-煅烧法制备。具体步骤如下:在室温下依次准确称取12 mmol CoSO4·7H2O和40 mmol CO(NH2)2,并溶于200 mL去离子水。充分溶解后将混合溶液转移至500 mL聚四氟乙烯内衬的水热釜中,然后在120℃下水热反应18 h。水热反应生成的沉淀(紫红色)以真空抽滤的方式分离出来,并用去离子水反复清洗。清洗后的沉淀在60℃干燥箱内干燥3 h,随后取出研磨成粉,即为催化剂前驱体。将前驱体在马弗炉中以2℃·min-1的速率升温至450℃后焙烧2 h,获得最终产物。
Co3O4-B采用化学浴沉积-煅烧法制备。具体步骤如下:在25℃水浴下将40 mmol CoSO4·7H2O溶于400 mL去离子水,得到暗红色均一溶液。然后在强烈的磁力搅拌下迅速加入10 mL浓氨水(25%~28%)并继续搅拌4 h,期间明显观察到大量沉淀的产生。将这些沉淀(深绿色)以真空抽滤的方式分离出来,并用去离子水反复清洗。将清洗后的沉淀置于60℃干燥箱内干燥过夜,随后取出研磨成粉,即为催化剂前驱体。将前驱体放在马弗炉中,以2℃·min-1的速率升温至450℃后焙烧2 h,获得最终产物。
Co3O4-C采用草酸盐热解法制备。具体步骤如下:首先准确称取18 mmol Co(NO3)2·6H2O溶于100 mL乙二醇+50 mL水组成的混合溶液中,记为A;准确称取18 mmol H2C2O4·2H2O溶于100 mL乙二醇+50 mL水组成的混合溶液中,记为B。然后在强烈的磁力搅拌下将B快速倾倒入A中,并继续搅拌20 min以使其混合均匀。混合均匀后的暗红色浊液被转移至500 mL聚四氟乙烯内衬的水热釜中,然后在120℃下水热反应16 h。水热反应生成的沉淀(粉红色)以真空抽滤的方式分离出来,并依次用去离子水和无水乙醇反复清洗。将清洗后的沉淀在60℃下干燥3 h,然后收集作为催化剂前驱体。前驱体在马弗炉中以2℃·min-1的升温速率被加热至400℃后焙烧2 h,从而获得最终产物。
1.2.2 PMS浓度测定
PMS溶液浓度采用碘化钾分光光度法进行测定[15-17],取2.5 mL质量浓度为1.66 g·L-1的KI溶液(其中含有质量浓度0.6 g·L-1的 NaHCO3,用以防止 I-被O2氧化)与0.5 mL PMS溶液混合,然后通过紫外可见光分光光度计在波长352 nm处测定其吸光度,并根据标准曲线确定PMS浓度。实验中PMS初始浓度为0.6 mmol·L-1,加入催化剂后立即计时,并于给定时间点取样,所取样品中的催化剂颗粒经由0.22µm混合纤维素滤膜滤去。
1.2.3 催化剂表面羟基密度测定
催化材料表面羟基密度采用文献报道的饱和脱质子法进行测定[18-19],并根据实验具体情况对所用试剂浓度及用量进行了调整。具体步骤如下:称取催化剂约0.02 g置于100 mL锥形瓶中,加入20 mL浓度5 mmol·L-1的NaOH溶液,混合均匀后放入恒温振荡箱中,在30℃下连续振荡3 h以使反应充分,然后用0.22µm混合纤维素滤膜滤去催化剂颗粒。取滤液 10 mL 用 5 mmol·L-1稀 HNO3滴定(以甲基橙为指示剂),记录所用稀HNO3体积ΔV1,同时测定未加催化剂的NaOH溶液所消耗的稀HNO3体积ΔV0,则催化剂表面羟基密度D(mmol·g-1)由式1计算得出:D=4×(ΔV0-ΔV1)×0.005/m,其中m表示催化剂实际所称质量。
1.2.4 MB催化降解实验
实验中选取典型的水溶性阳离子型染料MB为降解模型,以验证Co3O4/PMS高级氧化体系对有机染料废水的处理能力。具体步骤如下:首先准确称取一定量催化剂,然后加入到MB溶液中,开启机械搅拌,使其充分混合50 min以达到吸附饱和,取样并测定达到吸附饱和时的MB溶液浓度。随后加入一定量PMS,继续搅拌并在给定的时间点取样,以测定不同反应时间下溶液中的MB浓度。MB溶液的初始质量浓度为10 mg·L-1,体积为500 mL;为避免反应温度对PMS活性的影响,所有降解实验均在25℃恒温水浴下进行;MB溶液质量浓度的测定采用分光光度法完成,每次取样3.5 mL,所取样品首先通过0.22µm尼龙滤膜过滤,然后立即加入0.15 mL浓度3 mol·L-1的NaNO2猝灭反应,最后测定其在500~700 nm波长下的最大吸光度值。MB降解率(η)通过式2进行计算:η=(ρ0-ρt)/ρ0×100%,其中 ρ0表示达到吸附饱和时溶液中MB的质量浓度;ρt表示反应进行到t min时溶液中MB的质量浓度。
2 结果与讨论
2.1 三种Co3O4材料活化PMS的性能对比
对3种Co3O4材料活化PMS的性能进行了对比,结果如图1所示。根据文献报道,PMS被活化的过程本质是其在催化剂作用下发生分解,进而产生具有更高氧化活性的中间体产物如·SO4-、·OH、1O2或·O2-等。因此一般可通过PMS分解速率大小来比较催化剂的活化性能强弱。图1a展示了加入不同催化剂后PMS浓度随时间的变化。未加入催化剂时,PMS保持了较高的稳定性,其浓度随时间变化较小,25 min后分解率仅为1.39%,其少量分解可能是由环境可见光的光活化所引起;在3种不同催化剂的存在下,25 min后PMS分解率分别为68.45%(Co3O4-A)、98.18%(Co3O4-B)和 4.71%(Co3O4-C),即其分解率表现出不同程度的升高。按一级反应动力学模型对不同活化体系中PMS浓度与时间的关系进行拟合,结果如图1b所示。可以看出,3种Co3O4催化剂存在时PMS的分解过程均符合一级反应动力学模型,反应速率常数分别为0.047 1 min-1(Co3O4-A)、0.217 4 min-1(Co3O4-B)和 0.003 7 min-1(Co3O4-C)。根据实验结果,不同制备方法获得的Co3O4对PMS的活化能力强弱依次为Co3O4-B>Co3O4-A>Co3O4-C。
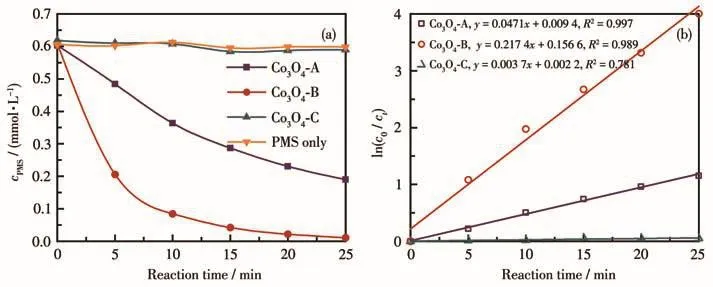
图1 不同制备方法所得Co3O4催化PMS分解的结果对比:(a)PMS分解过程;(b)一级反应动力学模型拟合结果Fig.1 Comparison of the results of PMS decomposition catalyzed by Co3O4 obtained from various methods:(a)decomposition process of PMS;(b)fitting results with the first-order kinetic model
2.2 材料的表征
2.2.1 XRD分析
图2为不同材料的XRD分析结果。图2a证明3种含钴前驱体材料分别为Co(CO3)0.5(OH)·0.11H2O、Co(OH)2和CoC2O4·H2O,且3种前驱体显示出不同的结晶度。因Co(OH)2是在室温下合成,故其结晶度最差。从图2b则可看出,3种Co3O4材料的衍射峰位置及相对强度均与标准卡片中的立方晶系Co3O4(PDF No.42-1467)相匹配,证明所得3种前驱体材料经历热分解后均完全转变为Co3O4。衍射图中没有观察到其它杂峰出现,表明材料纯度较高。从整体上看,3种钴氧化物中以Co3O4-B的衍射峰强度最低且峰型宽化,表明其结晶度相对最差,而Co3O4-A和Co3O4-C的衍射峰峰型尖锐,半峰宽更窄,表明它们的结晶度更高,晶粒发育更完善。结合前文的PMS活化性能分析结果,发现不同Co3O4材料对PMS的活化作用与其结晶度之间尽管表现出了一定的相关性,但对于结晶度相近的Co3O4-A和Co3O4-C而言,两者的催化活性却存在明显差异,这说明材料的结晶度并不是影响其催化活性的主要因素。

图2 前驱体(a)和煅烧产物(b)的XRD图Fig.2 XRD patterns of the precursors(a)and calcined products(b)
2.2.2 SEM分析
三种不同制备方法获得的Co3O4材料的SEM结果如图3所示。从低放大倍数的图片中可以看出,Co3O4-A呈绒球状,单个小球直径约5µm,由大量纤维状“绒毛”组成;Co3O4-B呈无规则颗粒状形貌,颗粒大小在500 nm以下;Co3O4-C则为纤维状,纤维长2~3µm,直径在500 nm以内。进一步观察高放大倍数电镜图片发现,Co3O4-A中的“绒毛”和Co3O4-C中的纤维实际由无数粒径更小的纳米颗粒堆叠而成,而Co3O4-B则由一些小的纳米片堆叠而成,亦即3种Co3O4材料均具有微/纳米多级结构。

图 3 Co3O4-A(a)、Co3O4-B(b)和 Co3O4-C(c)的 SEM 图Fig.3 SEM images of Co3O4-A(a),Co3O4-B(b),and Co3O4-C(c)
2.2.3 介孔比表面积分析
对于非均相催化过程而言,化学反应主要发生在催化剂表面,因此材料的催化性能与其比表面积之间存着密切的关系,即比表面积更大的催化材料能为反应过程提供更多的活性位点,通常表现出更好的催化效果。
图4展示了3种材料的N2吸附-脱附曲线及孔结构分析结果。从N2吸附-脱附曲线来看,3种材料的分析结果相似,均为典型的第Ⅳ类Langmuir吸附-脱附等温线;其吸附支与脱附支不重合,表明测试过程中发生了毛细管凝集现象,由此可推断出3种材料中均存在介孔结构。从图4b中也可以看出3种材料的孔径分布主要集中于2~50 nm之间;吸附支与脱附支之间形成的迟滞环为H3型,表明材料中的孔道为由层状粒子堆积而成的狭缝形孔,这与通过SEM观察所得的结果相一致。采用Barrett-Joyner-Halenda(BJH)算法由吸附支计算出材料的比表面积及孔结构参数列于表1,结果表明材料的催化性能与其比表面积之间存在一定的相关性,比表面积的差异可能是影响Co3O4催化活性的因素之一。
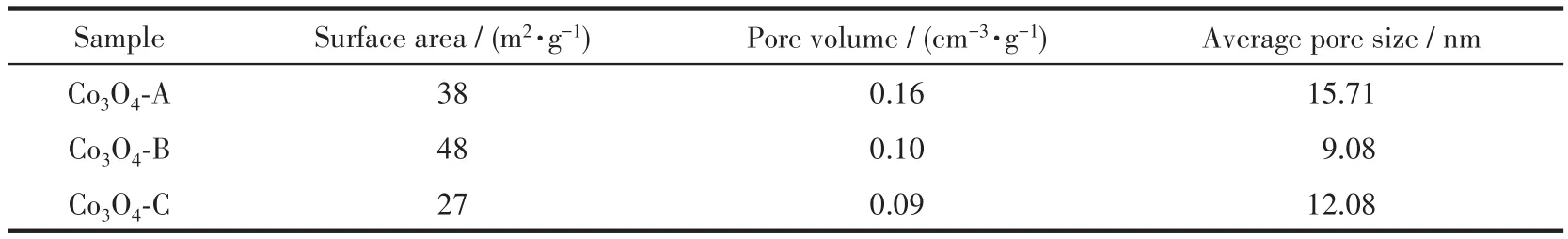
表1 三种Co3O4材料的比表面积及孔结构参数Table 1 Specific surface areas and pore structure parameters of three Co3O4 materials
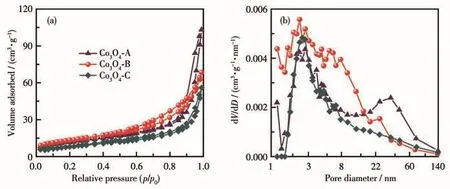
图4 三种Co3O4材料的N2吸附-脱附曲线(a)及孔径分布图(b)Fig.4 N2 adsorption-desorption curves(a)and pore size distribution plots(b)of three Co3O4 materials
2.2.4 氧空位分析
氧空位是一种晶体缺陷,由金属氧化物或其它含氧化合物中晶格氧原子的脱离而形成,具有较低的形成能,且其存在可能对催化剂的电子构型及物理化学性质产生影响。在非均相催化领域,合适的晶体表面缺陷通常有利于提升材料催化活性,如Wang等[20]发现不同的Co3O4对甲醛的催化降解活性与其表面氧空位密度存在明显的相关性,即氧空位密度更大的Co3O4表现出的催化活性也更高;Wang等[21]将铈锰复合氧化物用于催化脱苯,并认为复合氧化物具有比单独的氧化铈或氧化锰更高的氧空位浓度是其催化活性更高的原因之一。
为探究氧空位是否对不同Co3O4材料的PMS活化性能产生了影响,我们采用EPR法对3种Co3O4材料进行了氧空位的检测,测试结果如图5所示。在g=2.00处出现的信号证实在3种材料表面均存在氧空位缺陷,其相对强度分别为2 922、3 641和3 020,即表面氧空位密度大小为Co3O4-B>Co3O4-C>Co3O4-A,这与3种Co3O4材料的催化活性顺序不完全一致。该分析结果表明表面氧空位可能影响了Co3O4的催化活性,但这并非是决定其催化活性的主要因素。
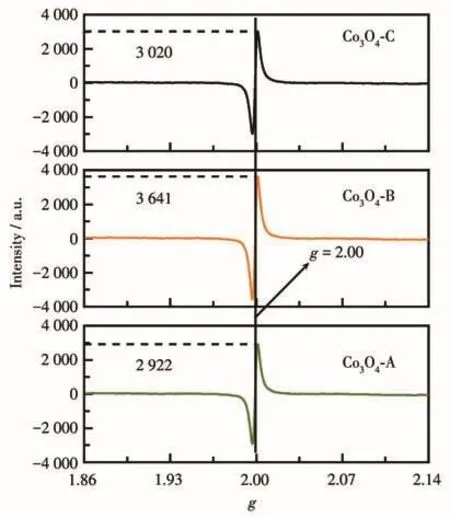
图5 EPR法检测的三种Co3O4材料的氧空位Fig.5 Oxygen vacancies from three Co3O4 materials detected by the EPR method
2.2.5 表面羟基密度
非均相催化剂表面的羟基基团由吸附的水分子在催化剂表面解离后产生,在PMS活化过程中可作为催化活性位点与PMS结合,形成M-OH-HSO5结构,这有助于增进PMS与催化剂的接触,进而促进对PMS的活化[3,22-23],因此非均相催化剂的催化活性受其表面羟基密度的影响也较大。我们采用饱和脱质子法[18-19]测得了3种Co3O4材料的表面羟基密度(D),并将该结果与对应的PMS活化反应速率常数(k)进行了对比,结果如图6a所示。可以看出催化剂表面羟基密度与在其活化作用下的PMS分解速率表现出显著的一致性,即催化剂的表面羟基密度越大,催化剂对PMS的活化性能越好。对PMS活化能力最好的Co3O4-B的表面羟基密度分别是Co3O4-A和Co3O4-C的5.83倍和13.59倍。该结果证实3种不同制备方法所得Co3O4对PMS活化性能的差异主要来自于其不同的表面羟基密度。
2.2.6 XPS分析
采用XPS对3种Co3O4材料中的主要元素及其表面化学价态进行了分析。图6b为全谱扫描结果。谱图显示主要检测到C1s、O1s和Co2p特征信号,其中位于284.1 eV处的C1s峰主要来源于仪器污染,该结果表明3种材料纯度较高,不含钴氧化物以外的杂质。Co2p的高分辨谱检测结果如图6c所示,3种材料的Co2p信号经分峰处理后均可拟合为位于780 eV的Co3+2p3/2峰、位于781.3 eV的Co2+2p3/2峰、位于795 eV的Co3+2p1/2峰和位于797 eV的Co2+2p1/2峰以及2个卫星峰。图6d为对O1s信号的分峰拟合结果,其均可分为3个小峰,且按结合能由低到高分别对应于晶格氧、表面羟基/吸附氧以及物理吸附氧[23-25],依次记为Oα、Oβ和Oγ。根据拟合峰面积计算出3种材料中Co2+与Co3+及不同化学态O的相对比例,列于表2。其中Co2+与Co3+比例与Co3O4中的理论比例1∶2不吻合,Co2+含量明显偏高,这主要与催化剂表面氧空位的存在相关;Co3O4-B的表面晶格氧含量明显偏低,与其低结晶度相对应;3种材料中Co3O4-B的Oβ含量比例最大,再次验证其具有更多的表面羟基。
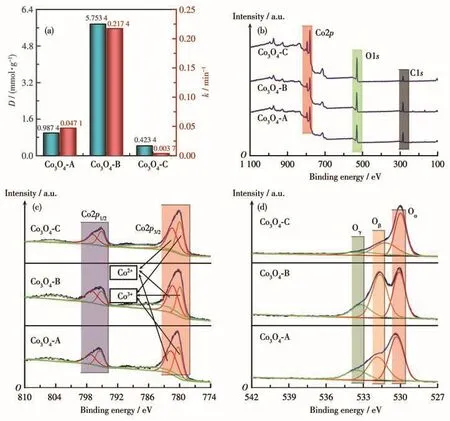
图6 三种Co3O4材料的表面羟基密度(a)及XPS分析结果:(b)总谱;(c)Co2p高分辨谱;(d)O1s高分辨谱Fig.6 Surface hydroxyl density(a)and the XPS analysis results of three Co3O4 materials:(b)full spectra;(c)high-resolution spectra of Co2p;(d)high-resolution spectra of O1s
2.3 MB催化降解实验
2.3.1 不同Co3O4/PMS活化体系下的MB降解实验结果
以MB溶液为降解模型,对3种不同制备方法所获Co3O4材料活化PMS处理模拟有机染料废水的性能进行评价和对比,结果示于图7。由图可见,在MB初始质量浓度10 mg·L-1、反应温度25℃、催化剂用量0.04 g·L-1、PMS投加量0.6 mmol·L-1的条件下,经单独的PMS处理50 min后,MB的η仅为9.32%;经Co3O4-C/PMS或Co3O4-A/PMS体系处理50 min后MB的η则分别可达31.55%或91.25%;而在Co3O4-B/PMS体系处理下MB的η仅需25 min即可达到100%。该结果表明3种催化剂均可提升PMS氧化降解MB的能力,且其中Co3O4-B表现出最佳的催化性能,其次为Co3O4-A,Co3O4-C则相对较差。不难看出Co3O4/PMS体系对MB溶液的降解效果与前文2.1小节给出的3种样品对PMS的活化能力具有一致性,这也说明本研究中Co3O4作为催化剂的作用是在PMS氧化降解MB的过程中活化PMS,产生活性中间体。
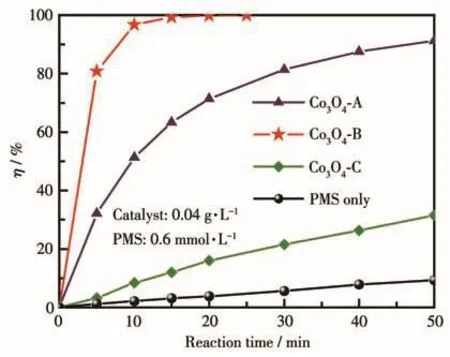
图7 三种Co3O4材料活化PMS降解MB的性能Fig.7 Performance of MB degradation with PMS activated by three Co3O4 materials
2.3.2 催化剂用量和PMS浓度的影响
在上述实验基础上,选取了具有最佳PMS活化性能的Co3O4-B催化剂做进一步研究。首先控制PMS浓度为0.6 mmol·L-1,对比了不同催化剂用量下的MB降解效果。从图8a可以看出,在反应时间25 min内,当催化剂用量为 0.01 g·L-1时,MB 的η 为89.27%,而催化剂用量增加至0.02 g·L-1以上时,其η可达98%以上,即MB的降解效果随催化剂用量的增加而得到提升。这是因为催化剂用量的增加可以为PMS活化提供更多的反应活性位点,促进更多活性中间体的产生,进而加速了其对MB分子的氧化降解。
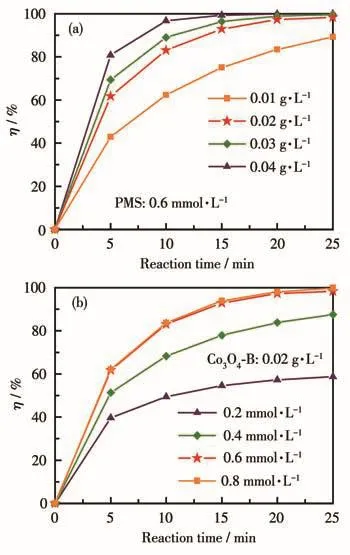
图8 催化剂用量(a)和PMS浓度(b)对Co3O4-B/PMS高级氧化体系降解MB的影响Fig.8 Effects of catalyst dosage(a)and PMS concentration(b)on the MB degradation in the Co3O4-B/PMS AOP system
同时也观察到,当催化剂用量为0.02 g·L-1时其对MB已有良好的降解效果,因此后续实验中将催化剂用量控制在0.02 g·L-1,对比不同PMS浓度对MB降解效果的影响,结果如图8b所示。当PMS初始浓度分别为0.2、0.4、0.6和0.8 mmol·L-1时,MB的η依次为58.72%、87.52%、98.33%和99.82%,说明PMS初始浓度的增加有助于提升MB降解效果。该降解效果的提升主要是由于PMS浓度增大引起的活性中间体的产生数量增加。另外,注意到当PMS浓度从 0.6 mmol·L-1增加至 0.8 mmol·L-1后,MB 降解效果并没有发生显著变化,这说明催化剂Co3O4-B浓度为0.02 g·L-1时PMS的饱和浓度不超过0.6 mmol·L-1,即由于催化剂表面的反应活性位点有限,当PMS 浓度达到0.6 mmol·L-1后,继续增大PMS浓度也不会促进更多数量活性中间体的产生。
综上,可以确定在25℃下由Co3O4-B/PMS高级氧化体系来处理10 mg·L-1MB溶液的最佳工艺参数:催化剂用量 0.02 g·L-1、PMS 投加量 0.6 mmol·L-1,在该条件下反应25 min内MB的η可达98.33%。
2.3.3 催化剂循环使用性能
使用后的Co3O4-B催化剂可采用简单的“过滤→洗涤→干燥”步骤进行回收,我们也对回收后的Co3O4-B催化剂的循环使用性能进行了测试,实验时催化剂用量和PMS投加量分别为0.04 g·L-1和0.6 mmol·L-1,实 验 结果见图 9。由图可 知 ,催化剂Co3O4-B的性能在前3次的循环使用时表现出明显的下降,MB的η从最初的100%依次下降至82.56%和67.43%,说明在循环使用过程中催化剂的表面活性位点存在一定的流失。幸运的是,在第4次循环使用时,MB的η略微提升到了69.02%,表明此时Co3O4-B的催化性能已经稳定,尽管与最初相比其催化性能下降明显,但仍表现出较好的催化降解MB效果。
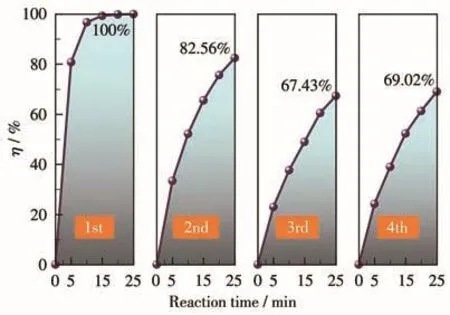
图9 催化剂Co3O4-B的循环使用性能Fig.9 Reuse-ability of the Co3O4-B catalyst
2.4 PMS活化机理分析
2.4.1 自由基猝灭实验
根据文献报道[26-29],TBA与·OH和·SO4-的反应速率常数分别为 k·OH=3.8×108~7.61×108L·mol-1·s-1,k·SO-4=4.0×105~9.1×105L·mol-1·s-1,即主要对反应体系中的·OH起猝灭作用;EtOH与·OH和·SO4-的反应 速 率 常 数 分 别 为 k·OH=1.2×109~2.8×109L·mol-1·s-1,k·SO-4=1.6×107~7.7×107L·mol-1·s-1,即对反应体系中的·OH和·SO4-均有良好的猝灭作用。因此,我们分别向反应体系加入了浓度为200 mmol·L-1的TBA或EtOH,通过对比MB降解效果的变化来间接推测PMS被活化时产生的自由基种类。从图10a所示结果可以看出,加入TBA后MB降解效果略有下降(η:98.33%→96.05%),这是由于PMS活化产生的·OH被TBA猝灭,由此可以推测PMS在Co3O4-B的活化下分解产生了少量·OH;而加入EtOH后,MB降解效果出现大幅下降(η:98.33%→77.09%),说明PMS在Co3O4-B的活化下产生了更多的·SO4-。
2.4.2 活性物种捕获实验
我们还采用EPR技术对反应体系中产生的活性物种进行直接检测,实验时催化剂用量0.04 g·L-1、PMS投加量0.6 mmol·L-1,测试结果示于图10b~10d。由图可知,催化反应发生2 min后,在Co3O4-B/PMS体系中同时检测到了DMPO-·SO4-、DMPO-·OH、DMPO-·O2-和TEMP-1O2四种加合物的特征信号峰,证明该体系中同时存在·SO4-、·OH和·O2-三种自由基型活性物种以及1O2非自由基型活性物种;而当反应进行到8 min时,所有特征信号峰强度均明显增强,表明体系中各种活性物种的浓度均存在随时间累积而增加的趋势。
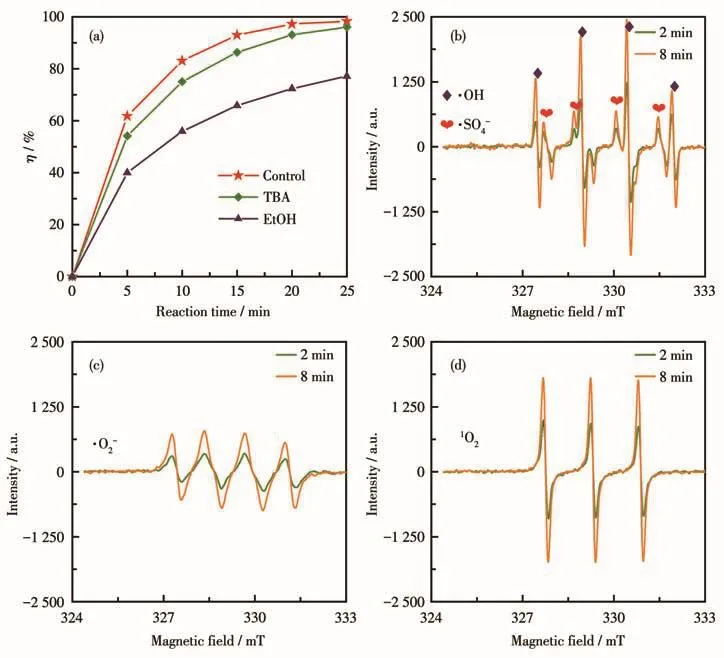
图10 Co3O4-B/PMS高级氧化体系中氧化活性物种鉴定结果:(a)自由基猝灭实验结果;(b~d)EPR检测结果Fig.10 Reactive oxygen species identification results in the Co3O4-B/PMS AOP system:(a)results of quenching experiments;(b-d)results of EPR detection
2.4.3 Co3O4-B/PMS体系降解MB溶液的机理
基于自由基猝灭实验和EPR实验结果,提出Co3O4-B/PMS体系降解MB的主要机理如下:
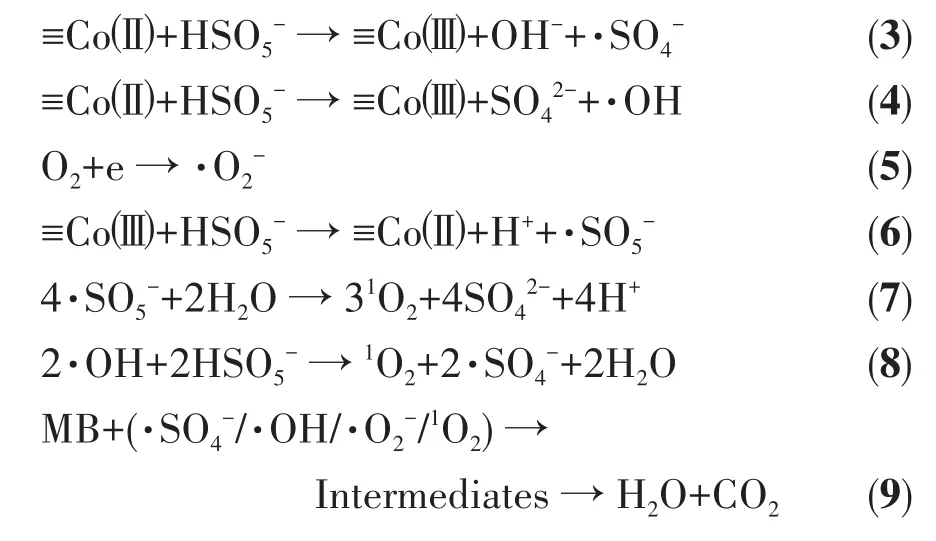
3 结 论
采用尿素水热-煅烧法、化学浴沉积-煅烧法和草酸盐热解法制得3种具有不同形貌的Co3O4材料,其中化学浴沉积-煅烧法所得产物Co3O4-B表现出最为优异的催化活化PMS的性能。这得益于该材料具有最大的比表面积、最高的表面氧空位浓度和表面羟基密度,尤以表面羟基密度贡献最为显著。通过单因素实验得出,在25℃下利用Co3O4-B/PMS体系处理10 mg·L-1MB溶液的最佳工艺参数为催化剂用量 0.02 g·L-1、PMS投加量 0.6 mmol·L-1,在此条件下反应25 min,MB的降解率可达98.33%。通过自由基猝灭实验证明Co3O4-B活化PMS产生的主要活性物种为·SO4-和少量的·OH,而进一步的EPR测试则表明Co3O4-B/PMS体系中还存在·O2-和1O2。
猜你喜欢
钛工业进展(2022年6期)2023-01-13
材料与冶金学报(2022年2期)2022-08-10
科学与财富(2021年33期)2021-05-10
作文成功之路·小学版(2020年6期)2020-07-27
作文成功之路·小学版(2020年5期)2020-06-11
中国化肥信息(2019年6期)2019-08-27
石油石化绿色低碳(2019年6期)2019-02-13
浙江大学学报(工学版)(2016年11期)2016-06-05
Coco薇(2016年2期)2016-03-22
