
C2 链烃在热解/气化中的碳链裂解机理及速率常数计算研究
2023-02-07 08:01朱中旭唐烽金余其陈思雨马家瑜
燃料化学学报 2023年2期
朱中旭,唐烽,金余其,陈思雨,马家瑜
(浙江大学 能源清洁利用国家重点实验室,浙江 杭州 310027)
热解/气化工艺可以减少污染物排放[1,2]、实现碳固定[3]并且通过对气相产物的利用来提高能源效率,因此,在许多发达国家的能源供应中有着重要应用[4-6]。目前,在工业应用中比较常见的气化剂有:空气、氧气以及水蒸气。此外,CO2作为一种纯净的O 供体[7],在作为气化剂方面也极具潜力,已有文献报道了其在气化过程中的作用[8-10]。气化剂种类繁多,但其本质都是通过产生H/OH/O自由基来与物料进行反应。
近年来,碳基燃料在气化领域取得了巨大的进展,但是焦油的存在阻碍了该工艺的清洁高效利用[11,12]。生活垃圾气化过程中的焦油主要来源于物料中的塑料组分,除了PS 和PET 产生的初级焦油具有芳香结构外,其余绝大部分为链烃结构[13],其中,轻质链烃化合物(C2-C4) 是形成焦油的关键,这类化合物可以通过脱氢后加成或者Diels-Alder 反应进一步环化和芳香化形成芳香化合物来促进焦油的产生。
为了理解轻质链烃化合物在热解/气化过程中的裂解机理,表征基本反应是必要的。目前,已有学者利用激波管反应器或者交叉分子束实验装置及配套的检测设备对轻质链烃化合物与H[14,15]、O[16,17]、OH[18,19]的反应进行了动力学实验,对通道产率进行了速率常数的表征。然而,由于链烃化合物的反应时间短[20],并且存在多种反应路径,实验的方式很难对单一反应过程进行精准的检测和分析。此外调查NIST 数据库[21]可以发现,针对轻质链烃化合物裂解的实验拟合结果主要集中在低温范围内,而理论计算虽对温度范围进行了扩展,但在C2-C4链烃化合物裂解,尤其是C2链烃碳链断裂方面上的研究依旧不够完善。
目前,Gaussian 软件已被广泛应用于有机化合物的精确计算[22-25],并通过和相关试验结果的比较验证了计算的准确性。本研究基于Gaussian 及其配套软件从微观的角度对结构最为简单的链烃类物质(乙烷、乙烯和乙炔)进行分析,对其可能的自由基反应位点进行了预测,并研究了其自身热裂解过程或者在自由基(包括O、H、OH)及水分子作用下的碳链断裂过程,探索了在热解/气化过程中鲜有研究的反应路径,完善了C2链烃化合物的裂解机理。在此过程中,比较了相关反应的速率常数随温度的变化情况,以定量化的方式分析了上述轻质链烃的裂解路径,扩充了理论数据。
1 计算原理及方法
本研究利用了Gaussian 16[26]、Multiwfn 程序[27-29]对自由基在乙烷、乙烯和乙炔上的可能进攻位点进行预测,并利用分子视图软件VMD[30]对结果进行可视化。随后,使用Gaussian 16[26]、以及Shermo软件[31]对C2系列链烃化合物的反应过程进行模拟计算,并利用GaussView 6.0 观察分子结构。
首先,应用Gaussian 16 对DFT 方法B3LYP/6-311G++(d,p)基组下的乙烷、乙烯和乙炔进行结构优化,然后将计算结果导入Multiwfn 中使用主功能12:分子表面的定量分析,分别算出在电子密度为0.001 a.u.等值面上的静电势(ESP)以及平均局部离子化能(ALIE),有关ESP 和ALIE 的定义可参考付蓉等的文章[32],此处不做详细介绍。
其次,使用Gaussian 16 的TS 功能对B3LYP/6-31G(d)基组下的过渡态进行计算,频率验证如附表1 所示,并对过渡态结果进行虚频的唯一性验证以及内禀反应坐标(IRC)计算以保证过渡态猜测的合理性。在有关过渡态及后续对于B3LYP泛函的计算中,均添加了色散校正。为了进一步得到更高精度的计算结果,耦合簇理论的应用必不可少[33,34],但是完整的耦合方法计算量太大,在实际计算过程中大多进行了相应的简化[35,36]。
目前,比较常用的是非迭代三重激发态的耦合簇CCSD (T)与Dunning 相关一致基组[37]相结合的计算组合模式。本研究在对反应过程中所涉及到的反应物、过渡态及产物进行了相应的优化和振动分析后,对这些化合物在更高的CCSD(T)/ccpVTZ 精度下进行单点能计算。然后,将计算结果导入Shermo 软件进行热物性特性参数的计算。在此过程中,零点能量(ZPE)、内能(U)、熵(S)采用的校正因子分别为0.9887、1.0102 及1.0161[38]。若涉及到多步反应,还需先在B3LYP/6-311G++(d,p)基组下计算每一步相关化合物及过渡态的自由能,并结合相应的能垒图判断整个反应过程的决速步。然后,再在CCSD(T)/cc-pVTZ 下计算反应决速步所涉及的反应物、最终产物以及对应的过渡态的单点能,并利用Shermo 完成热物性参数的计算。
通过上述方法计算得到的各个化合物的热物性参数可以代入到公式(1)中进行反应速率常数的计算。

式中,K(T)是基于Skodje-Truhlar 方法计算得到的量子力学隧道效应的透射系数[39],σ为反应对称系数,c0是p0/kBT(p0为0.1 MPa),ΔG(T,s0) 是根据反应决速步理论[40]确定的过渡态与反应物之间的自由能垒,s0是决速步所对应过渡态在反应坐标系上的位置,T是温度,kB、h分别是玻尔兹曼常数和普朗克常数。
2 结果与讨论
2.1 C2 系列链烃化合物的反应位点预测
分子表面静电势ESP 常被用于研究分子间的相互作用[41],由于在化学反应初期不同分子及基团之间通过静电吸引而互相接近,因此,通过对ESP 的定量研究可以预测反应物之间的相对位置,为后续的反应位点预测提供基础。图1 给出了乙烷、乙烯及乙炔的表面静电势,蓝色区域代表电势相对较低区域,易吸引亲电试剂(如自由基以及H2O 中的H)的靠近;红色区域表示电势相对较高区域,易吸引亲核试剂(如H2O 中的O)的靠近。其中,黄色小球为静电势极大值点,青色小球为极小值点(数据详见附表2、3)。由图1 可见,对于C2烃类而言,静电势的极大值点多数位于H 原子附近的分子表面上,而极小值点则多数位于C 原子附近,这主要是因为H 原子的电负性小于C 原子所致。由于C2链烃的静电势有正有负,这就会使得H2O 分子在靠近C2链烃的过程中受到静电力作用而自身极化,进而共价键断裂产生自由基参与到后续的反应之中,因此,本研究的反应都可以归结为自由基反应。此外,随着不饱和度的增加,静电势较低区域逐渐集中到C-C 键附近,这是由于π 键的形成导致碳碳双键及三键周围产生π 电子云,降低了附近区域的静电势。观察不同分子上的极小值点也可以发现,乙烯和乙炔的极小值点几乎相同并且均小于乙烷分子上的极小值(-0.004 a.u.),因此,O、H、OH 自由基更容易聚集在乙烯和乙炔的不饱和键附近增加彼此之间反应的几率。
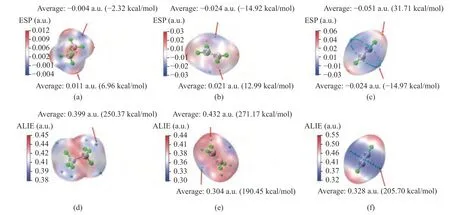
图1 C2 链烃化合物在电子密度为0.001 a.u.等值面上的静电势(ESP)及平均局部离子化能(ALIE)分布图Figure 1 Distribution of electrostatic potential and average local ionization energy of C2 chain hydrocarbons on the isosurface of electron density of 0.001 a.u.((a),(b),(c) are ESP;(d),(e),(f) are ALIE)
平均局部离子化能(ALIE)可以考察分子中的电子在局部位置上的被束缚程度,其数值越低表明电子在某处的束缚越小,相应的也越容易参与亲电或自由基反应[32],此前已有文献通过ALIE 对不同化合物的反应位点进行了预测[27,42-44]。在本研究中,乙烷、乙烯及乙炔的ALIE 分布如图1 所示,其中,蓝色代表数值较低区域;红色代表数值较高区域,青色小球为极小值点。从图1 中可以看出,乙烷的极小值点均匀分布在H 原子周围,表明H 原子是最有可能反应的活性位点,然而结合其ESP 分析可以发现,这些位点附近反而是ESP 的极大值点,是不太容易吸引自由基的位置,鉴于乙烷的ESP 极小值点与ALIE 极小值点位置截然相反,因此,乙烷的H 原子以及C 原子都有可能是基团进攻的反应位点。而乙烯和乙炔的ALIE 最小极值点则与其相应的ESP 极小值点保持了良好的一致性,因此,C 原子是自由基进攻乙烯和乙炔的主要位点。
2.2 C2 系列链烃化合物的反应路径
鉴于通过O/H/OH 自由基进行简单的H 夺取来改变原有结构的反应路径众多[21],但是并没有实现真正意义上的碳链断裂,因此,本工作主要针对碳链断裂的反应进行研究,基于上节对于反应位点的预测结果,有关乙烷裂解的反应如下所示:
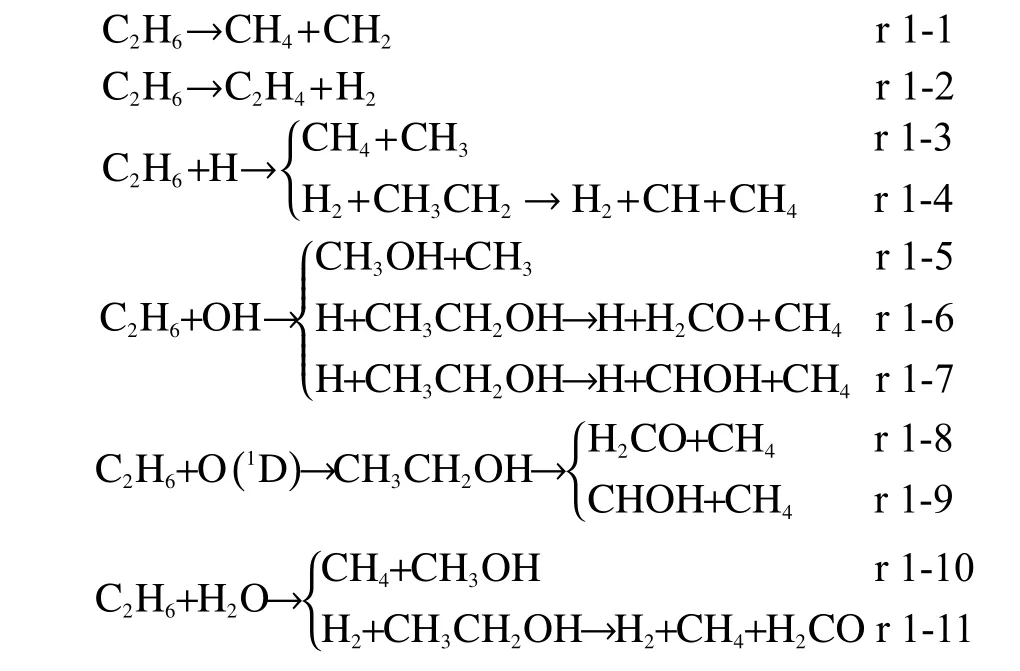
结合附图1 展示的过渡态结构来看,乙烷的热裂解是由于乙烷上的甲基发生旋转,使得甲基之间的排斥力增大从而促使C-C 键断裂,在此过程中伴随着H 转移产生CH4和CH2(路径r 1-1);也可以由甲基上的氢原子相互吸引生成H2和C2H4(路径r 1-2)。当H 自由基与乙烷反应时,H 自由基进攻C 原子促使乙烷碳链断裂并产生甲烷和甲基(路径r 1-3);也可以进攻甲基上的H 原子生成不稳定的CH3CH2后伴随着H 转移进一步断开C-C 键形成CH4和CH(路径r 1-4)。
目前在NIST 数据库[21]中关于OH 自由基与乙烷的研究主要集中在羟基对于乙烷上不同位置H 原子的夺取,而鲜少有关于其碳链断裂的相关报道,因此,本研究提出了几条可能路径如r 1-5至r 1-7 所示:路径r 1-5 类似于路径r 1-3,羟基通过进攻C 原子来夺取甲基,其产物为甲醇和甲基;而路径r 1-6 和路径r 1-7 则是在OH 自由基取代H原子之后,中间产物(乙醇)进一步分解为CH4和H2CO(路径r 1-6)或者CH4和CHOH(路径r 1-7),路径的不同是由于乙醇分解过程中不同位置的H 原子转移至甲基上所致。
类似地,目前有关乙烷和O 自由基反应的研究报道也主要集中在夺H[21],虽然已有文献报道了乙烷在O 自由基作用下的可能裂解反应,但均未就相关反应的速率常数随温度的变化情况进行研究,并且大部分研究都集中在三重态氧与乙烷的反应[45-48],因此,本研究参考前人的研究提出了两条可能的裂解路径(路径r 1-8 与r 1-9)并在此基础上研究了单重态氧与乙烷反应的速率常数。为促使C-C 键断裂,O 自由基首先进攻乙烷甲基上的H原子形成羟基,随后羟基与乙烷基结合生成乙醇,最后进一步裂解为CH4和H2CO(路径r 1-8)或者CH4和CHOH(路径r 1-9)。值得注意的是,在单重态O 原子参与反应时,由于该O 原子能量较高,乙醇的形成可看作是一个自发反应(图2 所示),该结果与Sun等[48]的计算结果一致。
水分子对乙烷的裂解也是通过夺取基团来实现,在路径r 1-10 中,水分子逐渐靠近乙烷导致C-C 键断裂,断裂形成的甲基又进一步增强了其与水分子中H 自由基以及OH 自由基之间的吸引,最终产生了甲烷和甲醇;而在路径r 1-11 中,水分子中的H 原子首先与甲基上的H 原子结合释放H2,而羟基与乙烷基结合形成乙醇,产生的乙醇再进一步通过裂解生成CH4和H2CO(即路径r 1-6),当然乙醇也可以沿路径r 1-7 的过程进行分解,但由于反应类似且乙醇分解的反应能垒没有水分子进攻H 原子高,此处不再详细介绍。
目前,有关乙烯和乙炔的热裂解反应都集中在H 脱除[21]的研究上,没有关于碳链断裂的报道,这可能是因为相比起烷烃,烯烃和炔烃在不饱和键处更难实现自身的裂解。由于C2系列化合物是最短的链烃其裂解难度本来就相对较大并且不饱和键处的断裂难度较高,在本研究所用方法下并没有找到合适的裂解路径,因此,在后续内容中不涉及有关乙烯以及乙炔的自身裂解路径,讨论核心将主要围绕乙烯和乙炔与自由基以及水分子的反应展开,乙烯裂解的相关反应如下所示:
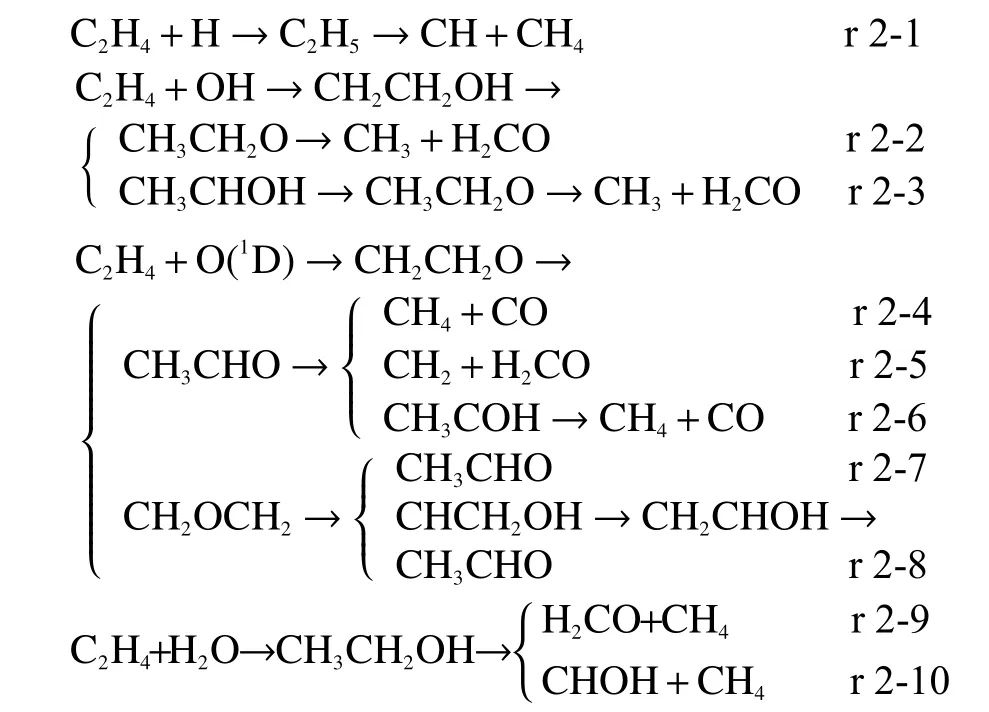
结合附图2 的过渡态结构可以发现,O/H/OH 自由基进攻乙烯C 原子实现对不饱和双键的加成,并且在经历一系列的旋转和结构重排后促使中间产物的进一步裂解。其中,H 自由基可与乙烯结合生成C2H5,随后伴随H 转移产生CH4和CH(路径r 2-1)。而当OH 自由基加成到乙烯之后,则主要通过内部H 转移来生成相对不稳定的CH3CH2O 促使C-C 键发生断裂,根据H 转移位置及步骤的不同有r 2-2 以及r 2-3 两条可能路径。
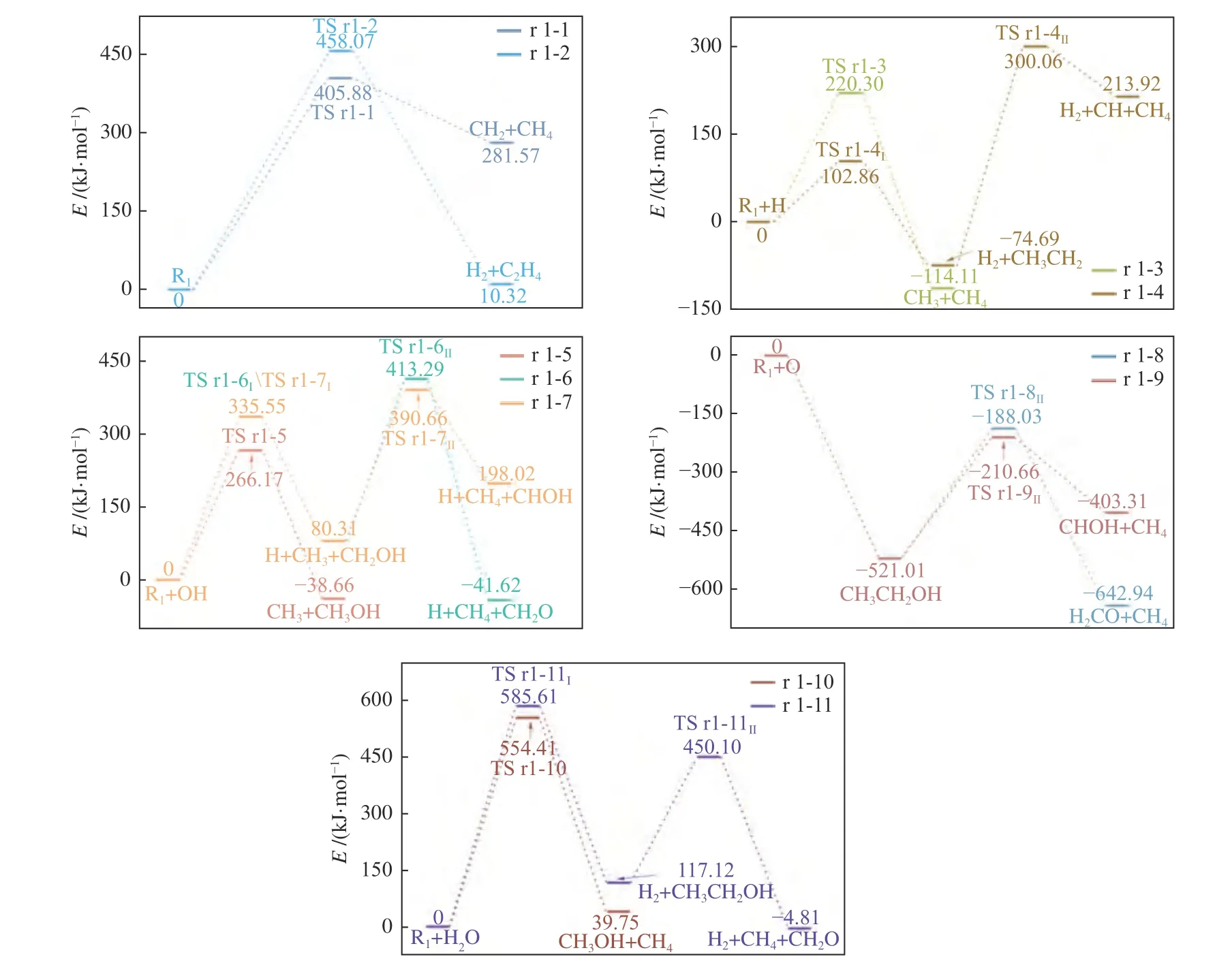
图2 乙烷在不同路径下的反应能垒变化Figure 2 Change of reaction energy barrier of ethane under different paths
对于O 自由基而言,本研究考虑了单重态O 的相关反应。根据图3 可以发现,在C2H4+O 生成CH2OCH2或者CH3CHO 的两步反应过程中,本研究并没有提供过渡态结构,这主要是因为第一步反应在B3LYP 方法上无法得到过渡态[49],而第二步计算所得过渡态能量处于反应物与生成物之间,因此,上述过程可以看成是自发反应,该结果与Talotta 等[50]的计算结果一致。由于在单重态势能面上CH2CH2O 是一个不稳定的中间产物并且会自发异构化[49],本研究将CH2CH2O 近似看成过渡态来简化反应路径,后续的讨论将重点围绕异构化产物乙醛以及环氧乙烷的反应展开,也不会影响整体的速率常数计算。
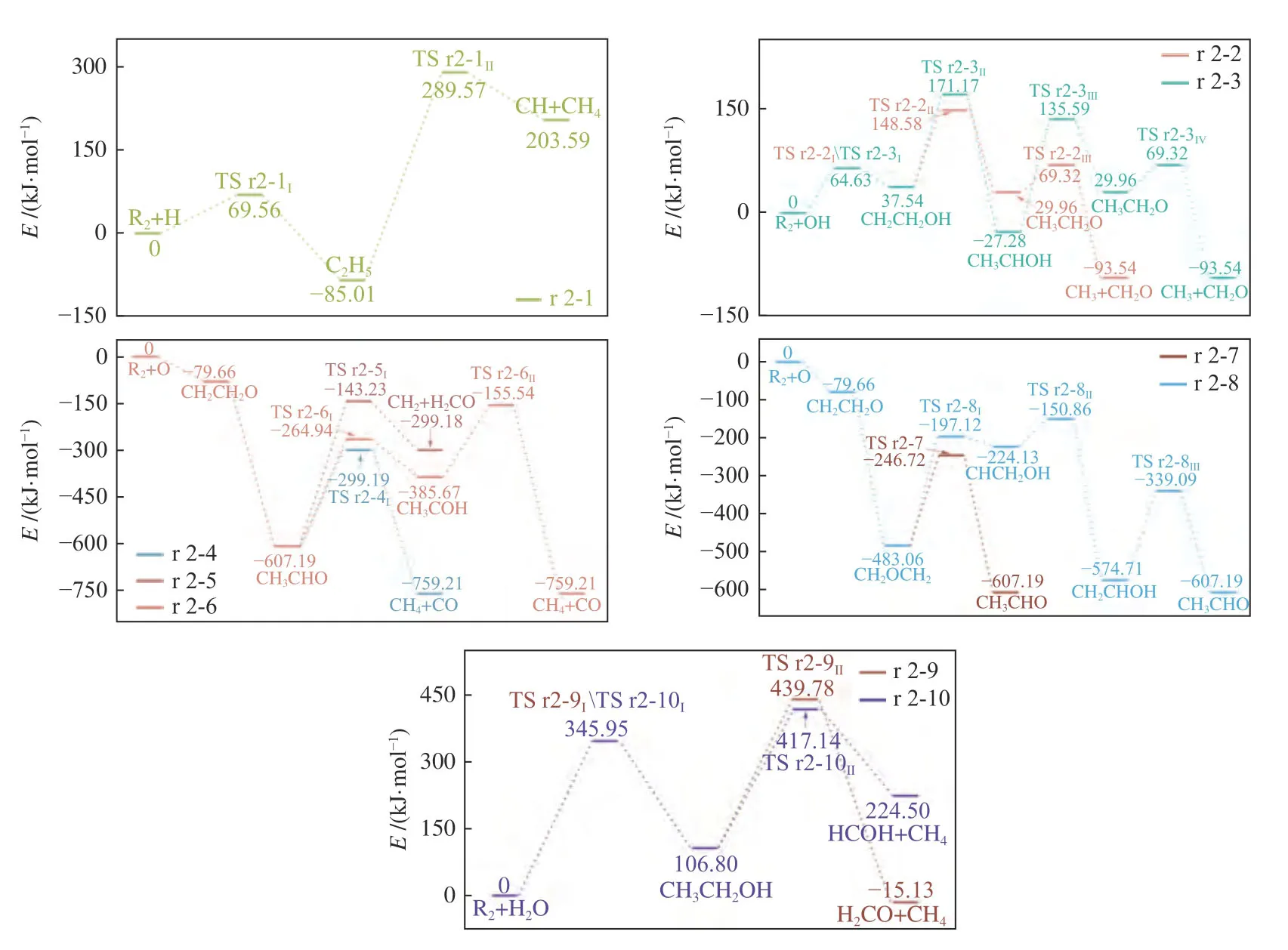
图3 乙烯在不同路径下的反应能垒变化Figure 3 Change of reaction energy barrier of ethylene under different paths
当乙烯与O 自由基反应生成乙醛之后主要可通过三条路径来实现碳链的断裂。在路径r 2-4中,甲基旋转带动乙醛裂解并伴随着H 转移形成CH4和CO;类似地,在路径r 2-5 中,CH3CHO 也是通过裂解以及H 转移形成甲醛以及亚甲基;而在路径r 2-6 中则是先通过内部H 转移产生CH3COH,然后碳链进一步断裂并伴随H 转移形成稳定的CH4以及CO。环氧乙烷则主要是通过异构化形成CH3CHO 后再进行裂解,其中,路径r 2-7是通过H 转移异构化为CH3CHO;而路径r 2-8 则是通过H 转移异构化生成CHCH2OH 之后先通过OH 转移生成CH2CHOH,再通过H 转移形成CH3CHO。
此外,当乙烯与水分子反应时,主要是通过与水分子裂解产生的H/OH 自由基进行加成反应生成CH3CH2OH,随后再进一步裂解为CH4和H2CO(路径r 2-9)或者CH4和CHOH(路径r 2-10)。
与乙烯不同的是,当乙炔与H 原子反应时,产物依旧保留不利于自身热裂解的不饱和键,Tsang 等[51]总结了多种乙炔与H 自由基反应的路径,但这些反应仅能实现H 的加成或者H 的脱除,而无法将乙炔裂解,此外在计算过程中使用的基组精度有限,未能找到合适的裂解路径,因此,本研究也不考虑乙炔在H 自由基作用下的裂解反应。下文将围绕乙炔与OH/O 自由基以及水分子的加成反应路径展开讨论,反应路径如下所示:
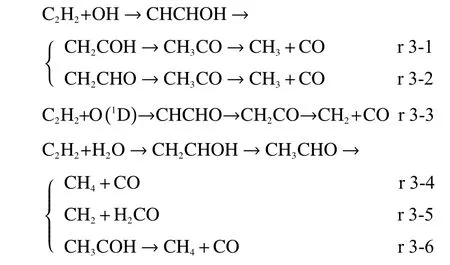
根据附图3 的过渡态结构,乙炔与OH 自由基首先进行加成反应生成CHCHOH,随后通过两次内部H 转移产生CH3CO,最后在甲基旋转的带动下碳链断裂产生CH3和CO,根据H 转移的顺序不同,有路径r 3-1 和r 3-2两种过程。
理论计算表明为保证乙炔断裂,乙炔与O 自由基首先加成产生CHCHO[23,52,53]。与乙烯类似,加成反应在B3LYP 方法上无法在单重态势能面上得到过渡态[52],该反应可看作是自发进行(图4 所示),本实验后续讨论将从CHCHO 开始。在路径r 3-3 中,当乙炔与O 自由基加成生成CHCHO 之后,将通过内部H 转移生成CH2CO,然后CH2CO进一步分解为CH2与CO。
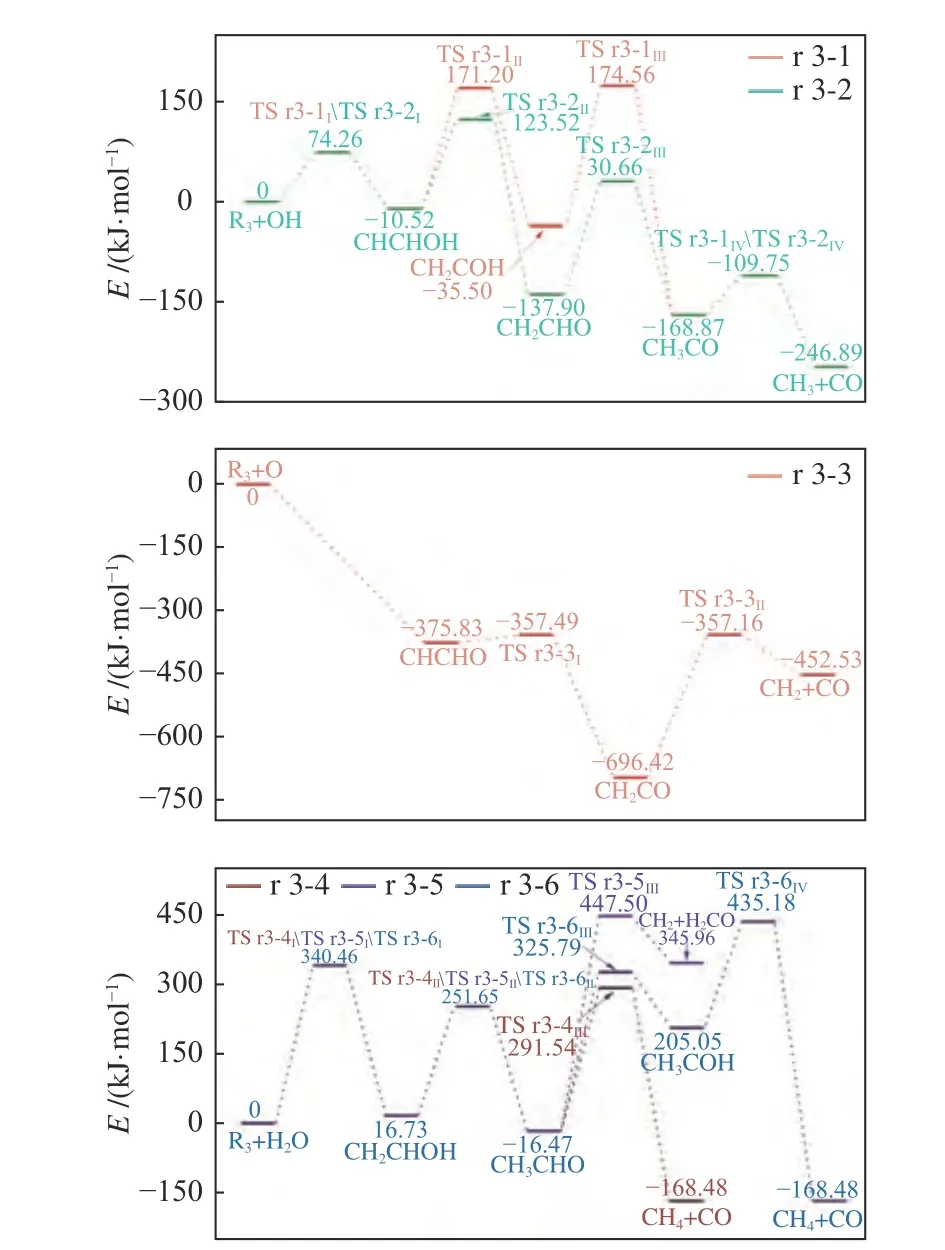
图4 乙炔在不同路径下的反应能垒变化Figure 4 Change of reaction energy barrier of acetylene under different paths
同样地,待水分子与乙炔加成生成CH2CHOH后,通过内部H 转移产生乙醛,再分解产生CH4和CO(路径r 3-4)或者CH2和H2CO(路径r 3-5);乙醛也可以通过内部H 转移生成CH3COH 之后再进一步裂解为CH4和CO(路径r 3-6)。
2.3 C2 系列链烃化合物的反应动力学
乙烷自身裂解的化学反应速率常数随温度的变化情况如图5 所示,从图5 可以看出,温度对速率常数的影响非常大,当温度从600 K 升至1500 K,路径r 1-1 和r 1-2 的反应速率常数分别有23 个和26 个数量级的提升。但在相同温度下,路径r 1-1更容易进行,相应的速率常数要比路径r 1-2 高出2-5 个数量级。这说明相比起C-H 键断裂产生H2,乙烷更容易实现C-C 键的断裂。对比已有文献数据(附表4)可以发现,路径r 1-2 的速率常数计算结果与Ge 等[54]计算结果处于同一数量级。目前,已有乙烷自身裂解生成甲基的反应速率常数研究[55,56],而路径r 1-1 则是探究了甲基的进一步转换。因此,路径r 1-1 在700-1500 K 的速率常数要比文献记录的同温度下的结果低至少2 至8 个数量级,计算结果表明,对于乙烷而言,其自身的热裂解相对比较容易,但较难通过基团之间的H 转移来产生甲烷。
图5 也展示了H 自由基与乙烷反应从而促使其裂解的化学反应速率常数,可以看出,随着温度的升高速率常数逐步上升。其中,路径r 1-3 是与H自由基反应的最佳路径,其速率常数从600 K 的1.01 × 10-3L/(mol·s)增至1500 K的2.40 × 105L/(mol·s),均高于同温度下路径r 1-4 的速率常数。但随着温度升高,两条路径之间的速率常数差距也在逐步缩小,从600 K 的15 个数量级减少到1500 K 时的两个数量级。结合能垒图(图2)来看,路径r 1-4的决速步是C2H5的裂解反应,温度的升高对该步骤的促进效果明显。Back 等[57]根据乙烷热分解的实验结果估算了823-983 K 乙烷在H 自由基作用下裂解的反应速率常数,但由于反应压力的不同以及实验测量精度有限,路径r 1-3 的速率常数比估算结果要高出三个数量级。
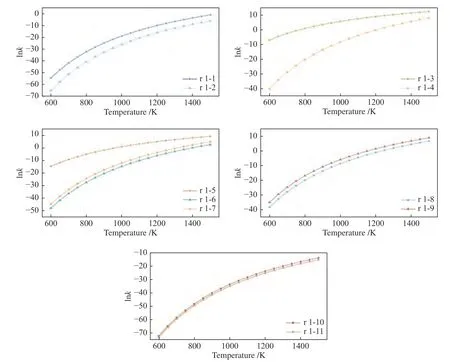
图5 乙烷在不同路径下的反应速率常数Figure 5 Reaction rate constants of ethane under different reaction paths
类似地,不同路径反应速率常数随温度上升而逐渐接近的现象在乙烷与羟基反应时也存在。如图5 所示,OH 自由基可以通过三种方式促进乙烷裂解,其中,单步反应r 1.5 的速率常数最大,从600 K 的4.08 × 10-7L/(mol·s)增至1500 K 的8.26 ×103L/(mol·s),在相同温度下,比路径r 1-6 和路径r 1-7 大了至少1-13 个数量级。从图5 也可以看出,三者之间的速率常数随着温度的升高而逐步接近,表明温度的升高有助于乙醇的分解(路径r 1-6及路径r 1-7 的决速步)。此外,对比r 1-6 和r 1-7的速率常数可以发现,在相同温度下r 1-7 的速率常数总比r 1-6 的高一个数量级,这表明,氢原子转移的间距也会影响裂解反应的快慢,H 转移终末两点位之间的距离越长会使得反应的能垒越高从而导致反应速率常数越小。
当O 自由基攻击乙烯导致其裂解时,根据决速步理论从图2 可以看出,两条可能路径(r 1-8和r 1-9)的决速步都是第二步,其中,路径r 1-9 的决速步能垒相对较低,在相同温度下的速率常数较高,这主要是因为上述两种路径的主要区别在于第二步的H 转移过程,反应难易程度取决于H 转移的能垒。此外,温度升高可使路径r 1-9 的速率常数显著增长,从600 K 升高到1500 K 的过程中提高了19 个数量级(2.37 × 10-17L/(mol·s) -8.90 ×102L/(mol·s))。
综合比较乙烷在H/OH/O 自由基作用下的速率常数,可以发现在600-1400 K,H 自由基对于乙烷的裂解效果最好,而O 自由基对于乙烷的裂解效果最差。另外,1500 K 时,路径r 1-9 的速率常数超过了路径r 1-5,并且从图5 的变化趋势来看,在1500 K 之后,若随着温度的继续升高,路径r 1-9 的速率常数增长率明显高于路径r 1-3,这就会使得路径r 1-9 的速率常数逐渐接近路径r 1-3,但由于此时温度超过了一般热处理设备的温度范围,讨论温度高于1500 K 的变化情况对实际工程指导意义不大。
此外,温度的升高同样能促进水分子对乙烷的裂解。当温度升至1500 K 时,反应速率常数均提高了25 个数量级,路径r 1-10 的速率常数比相同温度下路径r 1-11 的速率常数略大,但没有出现数量级的差异。但由于裂解路径均涉及到水分子的裂解,导致乙烷与水分子反应的能垒较高,其速率常数不如乙烷与自由基的反应。
图3 展示了乙烯在不同条件下的反应能垒。根据决速步理论[40],除了路径r 2-6 和r 2-8 的决速步是第三步,其他反应路径的决速步均为第二步。从乙烯加H 裂解的路径来看(图6),路径r 2-1的速率常数从600 K 的8.209 × 10-19L/(mol·s)增至1500 K 的2.915 × 102L/(mol·s)。将计算所得速率常数与Huynh 等[58]有关H 夺取反应(C2H4+H-C2H3+H2)的计算结果相比较(附表5),可以发现在600-1400 K 更容易发生夺H 反应,但当温度高于1500 K时,更倾向于路径r 2-1 的裂解反应。上述结果表明在高温下,H 自由基对于乙烯的裂解作用是不可忽视的。
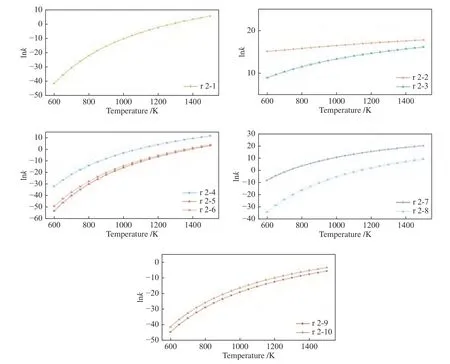
图6 乙烯在不同路径下的反应速率常数Figure 6 Reaction rate constants of ethylene under different reaction paths
OH 自由基与乙烯加成生成CH2CH2OH 后,可进一步通过内部H 转移有效促使碳链的断裂(路径r 2-2),相应的速率常数从600 K 的3.833 ×106L/(mol·s)增至1500 K的5.467 × 107L/(mol·s),比相同温度下路径r 2-3 的速率常数高出至多3 个数量级。结合路径r 2-3 的能垒图(图3)来看,相比起路径r 2-2,路径r 2-3 多了一步H 转移过程并且CH2CH2OH 通过H 转移形成CH3CHOH 的能垒更高,因此,实现该路径所需的能量将会增加,使得路径r 2-3 的速率常数较小,但随着温度的升高,速率常数之间的差距逐渐减小。与夺H 反应(C2H4+OH-C2H3+H2O)相比[19,59],路径r 2-2 随温度的变化趋势与其一致,但要低两个数量级。考虑到乙烯与OH 自由基反应的路径本身速率常数就较高,因此,羟基促使乙烯断裂也是一条重要的反应路径。
O 自由基参与乙烯裂解反应的速率常数主要取决于乙醛的裂解方式,其中,最容易进行的是路径r 2-4,相应的速率常速从600 K 的1.648 ×10-14L/(mol·s)增至1500 K的1.248 × 105L/(mol·s)。值得注意的是,虽然路径r 2-6 需要通过H 转移产生CH3COH 来实现加成产物的进一步分解,但其相应的速率常数虽然比相同温度下路径r 2-4 低4.6 个数量级但是仍比路径r 2-5 高出至多两个数量级。这说明速率常数的高低主要取决于路径能垒的大小,由于CH3CHO 与TS r 2-6Ⅱ之间的能垒始终低于其与TS r 2-5Ⅰ的能垒,因此路径r 2-6 的反应速率常数会更大。在相同温度下,路径r 2-4的速率常数同Li 等[60]的模拟结果相差较大(附表2),这也说明单重态氧需要更多的能量才能够促使乙烯断裂,因此,单重态氧对于乙烯的裂解作用只有在高温才会体现。此外,对于环氧乙烷转换为乙醛的反应路径而言,虽然路径r 2-8 是一条可行路径,但其速率常数在相同温度下比路径r 2-7的速率常数低了5-11 个数量级。
同样,综合比较乙烯在H/OH/O 自由基作用下的速率常数,路径r 2-2 也是图6 中最容易发生的路径,这充分表明,OH 自由基在乙烯的裂解中占有重要地位。与乙烷不同的是,在同温度下不同自由基对乙烯裂解的效果为:OH 自由基 > O 自由基 > H 自由基。此外,由于O 可以取代H 得到OH自由基[61-63],在氧化性气氛下乙烯裂解也是可行的。
当水分子和乙烯进行反应时,路径r 2-10 的速率常数在相同温度下始终比路径r 2-9 的高出1-2 个数量级。从前面的分析可知,两种反应路径的关键步骤都是第二步(乙醇分解过程)。与前文类似,该两条路径速率常数的大小取决于H 转移的位置,由于路径r 2-10 的转移位点之间距离更近,因此其速率常数始终高于路径r 2-9。
图7 展示了乙炔在不同反应条件下速率常数的分布情况。当乙炔在羟基作用下裂解时,路径r 3.2的速率常数在相同温度下始终比路径r 3-1 高1-2 个数量级,其速率常数从600 K 的9.206×10-1L/(mol·s)增至1500 K 的2.041×109L/(mol·s),从图4 中可以发现,虽然两条路径都是通过两次内部H 转移产生CH3CO 后导致碳链断裂,但是路径r 3-2 的能垒始终小于路径r 3-1,表明H 转移顺序的不同对于整体路径的能垒具有一定的影响,对于CHCHOH而言,羟基上的H 原子转移至间位碳原子比起羟基相连碳原子上的H 原子转移至另一碳原子上是更加容易的,而这一转换路径也导致后续H 转移的位移距离更近,能垒更小。路径r 3-2 的结果与Miller 等[64]的计算结果在1000-1100 K 具有同一数量级(附表6)。
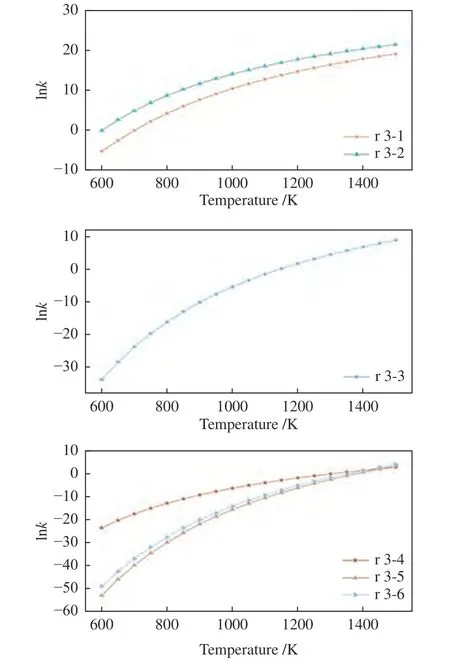
图7 乙炔在不同路径下的反应速率常数Figure 7 Reaction rate constants of acetylene under different reaction paths
当单重态氧参与乙炔的裂解反应时,反应速率常数随着温度的升高而增加。但与三重态氧的相关反应速率常数[65,66]相比,其在同温度下的速率常数则小了至少6-20 个数量级,该结果表明,对于乙炔而言单重态氧似乎较难与乙炔反应促使碳链断裂,因此,在实际反应过程中路径r 3-3 的发生可能性较小。
从水分子与乙炔反应的能垒分布情况(图4)来看,路径r 3-4 的决速步是第一步,路径r 3-5 是第三步而路径r 3-6 是第四步,其中,路径r 3-4 变化相对平缓,从600 K 的5.169×10-11L/(mol·s)增至1500 K 的1.479×101L/(mol·s),而路径r 3-5 和 r 3-6变化较为剧烈,分别增加了25 及23 个数量级。在600-1400 K,路径r 3-4 的速率常数是最大的,但当温度超过1400 K 之后,路径r 3-6 和路径r 3-5的速率常数也陆续超过了路径r 3-4。上述结果表明,相比起H2O 与C2H2的加成反应,温度对于后续裂解反应的促进作用更加明显。
综合比较乙炔裂解的各个路径可以发现,OH自由基对于乙炔的裂解效果最好,在相同温度下路径r 3-2 的速率常数最大。将路径r 3-2 的速率常数与路径r 2-2(乙烯在OH 自由基作用下的最佳裂解路径)的速率常数进行比较可以发现,当温度低于1200 K 时,路径r 2-2 的速率常数始终高于路径r 3-2(见附表7),表明此时OH 自由基更容易与乙烯反应;但当温度高于1200 K 后,路径r 3-2的速率常数明显更高,此时OH 自由基更倾向于同乙炔反应。根据各个路径的决速步可知,这主要是因为不同含氧基团在不同温度下的裂解难易程度发生了变化,其中,CH2CH2OH 裂解(路径r 2-2决速步)相比起CH2CHO 裂解(路径r 3-2 决速步)在低于1200 K 的环境内更容易发生,但由于CH2CH2OH 的裂解增速随温度的变化程度不如CH2CHO 剧烈,从而导致温度高于1200 K 后CH2CHO 更容易裂解。
此外,结合乙烷和乙烯的相关结论发现,对于C2系列链烃化合物而言,OH 自由基对不饱和烃的裂解效果最佳,而H 自由基对于饱和烃的裂解效果更好。由于水分子是上述两种自由基的供体,不难推断水蒸气的添加有利于C2碳氢化合物的裂解,因此,在热解/气化的实际工程应用过程中,在满足入炉物料热值要求的前提下可考虑适当保留其中的水分以促进C2链烃化合物的裂解。最后,本文所涉及路径的反应速率常数相关计算参数和结果均列在附表7、8 中。
3 结论
本研究采用Gaussian 及其配套软件从微观的角度对乙烷、乙烯和乙炔的热解/气化机理进行了探索,所得结果如下。
计算结果表明,自由基反应可能在乙烷的C 原子及H 原子上发生而对于乙烯和乙炔而言则主要集中在C 原子上,因此,H/OH/O 及水分子通过进攻乙烷中的甲基基团来实现碳键断裂,而对于乙烯及乙炔这两种不饱和烃则主要通过攻击双键或者三键位置进行加成后才能实现裂解。
当OH 与乙烯和乙炔进行加成后,中间产物的O-H 键相较于C-H 键更容易断裂;而当一个C 原子上同时存在H、OH 自由基时,羟基上的H 转移所需要的能量更少。另外在600-1200 K,OH 自由基更容易与乙烯反应;而在高于1200 K的环境下,OH 自由基更倾向于同乙炔反应。该结果也反映了不同含氧基团对于温度变化响应速度的差别,从中可以推断出醛类基团对于温度变化的响应速度优于醇类基团。
最后,碳链断裂过程伴随着H 转移,该步骤在C2系列链烃化合物的裂解过程尤其是在含有不饱和键结构的裂解过程中较为常见。另外,OH 自由基对不饱和烃的裂解效果最佳,而H 自由基对于饱和烃的裂解效果更好,因此,适量水蒸气的存在会对C2链烃化合物的裂解具有进一步的促进作用。
猜你喜欢
煤气与热力(2021年3期)2021-06-09
科普创作(2018年1期)2018-11-30
石油化工建设(2018年2期)2018-07-11
铜仁学院学报(2018年6期)2018-07-05
电源技术(2016年9期)2016-02-27
自动化博览(2014年9期)2014-02-28
自动化博览(2014年8期)2014-02-28
无机化学学报(2014年9期)2014-02-28
无机化学学报(2014年8期)2014-02-28
中国氯碱(2014年12期)2014-02-28
