
CMT1A current gene therapy approaches and promising biomarkers
2023-02-24 05:24MarinaStavrouKleopasKleopa
中国神经再生研究(英文版) 2023年7期
Marina Stavrou ,Kleopas A.Kleopa,
Abstract Charcot-Marie-Tooth neuropathies (CMT) constitute a group of common but highly heterogeneous,non-syndromic genetic disorders affecting predominantly the peripheral nervous system.CMT type 1A(CMT1A) is the most frequent type and accounts for almost~50% of all diagnosed CMT cases.CMT1A results from the duplication of the peripheral myelin protein 22 (PMP22) gene.Overexpression of PMP22 protein overloads the protein folding apparatus in Schwann cells and activates the unfolded protein response.This leads to Schwann cell apoptosis,dys-and de-myelination and secondary axonal degeneration,ultimately causing neurological disabilities.During the last decades,several different gene therapies have been developed to treat CMT1A.Almost all of them remain at the preclinical stage using CMT1A animal models overexpressing PMP22.The therapeutic goal is to achieve gene silencing,directly or indirectly,thereby reversing the CMT1A genetic mechanism allowing the recovery of myelination and prevention of axonal loss.As promising treatments are rapidly emerging,treatment-responsive and clinically relevant biomarkers are becoming necessary.These biomarkers and sensitive clinical evaluation tools will facilitate the design and successful completion of future clinical trials for CMT1A.
Key Words:axonal degeneration;biomarkers;Charcot-Marie-Tooth disease;gene therapy;inherited neuropathy;mouse models
Introduction
Charcot-Marie-Tooth neuropathy (CMT;also known as hereditary motor sensory neuropathy) encompasses a group of highly heterogeneous,nonsyndromic genetic disorders that predominantly affect the peripheral nervous system (PNS).CMT-related inherited neuropathies have an esti mated overall prevalence of around 1 in 2500 people (Skre,1974;Barreto et al.,2016).Heterogeneity of CMTs is related to their clinical features,mode of inheritance,CMT-causing genes and different mutations hosted within causative genes.Until today,more than 100 different CMT-causative genes have been identified,which have a wide range of functions,leading to different CMT subtypes and disease mechanisms (Stavrou et al.,2021a).
There is extremely high variability in the clinical features,age of onset and progression rate among different CMT types and even among individuals of the same family carrying the same mutation (Sevilla and Vilchez,2004).In most affected individuals,CMT symptoms become apparent between 5–25 years of age.In general,CMT patients show a progressive phenotype characterized by dysfunctional lower motor and sensory neurons (Rossor et al.,2012;Bansagi et al.,2017;Rudnik-Schöneborn,2020),which leads to a length-depended axonal degeneration (Vinci,2003).The clinical manifestations include symmetrical distal muscle weakness and atrophy,distal sensory loss in a stocking-glove distribution,and diminished or absent deep tendon reflexes.Additional features include kyphoscoliosis,foot drop and gait disturbance (resulting from weak ankle dorsiflexion),as well as claw toe andpes cavusdeformity (also known as high-arched feet) (Stavrou et al.,2021b).The phenotype typically appears in the lower limbs and then progressively affects more proximal leg and hand muscles (Rudnik-Schöneborn,2020).CMT patients may also experience nociceptive and neuropathic pain caused by joint deformities and small nerve fiber impairment,respectively (Peretti et al.,2022).
CMT type 1A (CMT1A;OMIM #118220) is the commonest type of CMT(accounting for~50% of all CMTs) and results from the duplication of the peripheral myelin protein 22 (PMP22) gene located on chromosome 17p11.2(Skre,1974).CMT1A is characterized by dys-and de-myelination,onion bulb formations,and reduced nerve conduction velocities (both motor and sensory) (Stavrou et al.,2021b).Although the exact CMT1A cellular pathomechanism still remains elusive,it is speculated that overexpressed PMP22 saturates and disturbs proteasomal degradation pathways causing perinuclear (Notterpek et al.,1999;Ryan et al.,2002) and cytoplasmic(Hanemann et al.,2000) accumulations of PMP22,generally attenuated proteasomal acti vity (Fortun et al.,2005),and endoplasmic reticulum stress(Khajavi et al.,2007).Furthermore,PMP22 overexpression overloads the protein folding apparatus and activates the unfolded protein response in Schwann cells,leading to cell apoptosis,impaired myelin function,and secondary axonal degeneration,which ultimately causes neurological disabilities.These effects are predicted to cause the pathological hallmarks of CMT1A,including dys-and de-myelination,remyelination,and onion bulb formations (Khajavi et al.,2007).The consequences of these features include problematic Schwann cell-axon interactions and impaired neurofilament structure.These,in turn,result in inadequate phosphorylation and increased packing density of neurofilaments as well as slowed axonal transport (de Waegh et al.,1992;Robaglia-Schlupp et al.,2002;Saporta et al.,2009).
To understand the disease pathogenesis as well as to test novel therapies,CMT1A pathology has been reproduced in different transgenic rodent models.These models either carry missense mutations (Suter et al.,1992;Valenti jn et al.,1992;Suh et al.,1997;Isaacs et al.,2000) (features summarized inTable 1) or extra copies of thePMP22gene (Huxley et al.,1996,1998;Magyar et al.,1996;Sereda et al.,1996;Perea et al.,2001;Boutary et al.,2021b)(features summarized inTable 2) and have been proven very valuable in the effort to develop CMT1A treatments.However,as illustrated inTables 1and2,the severity of phenotypes can vary among the models,some of which are more accurate to reflect the CMT1A pathological changes and course of progression,while others are more severe that can best reflect congenital or early-onset severe demyelinating neuropathies.This should be taken into consideration when assessing the validity of treatment results in different models,along with the ti ming of intervention.
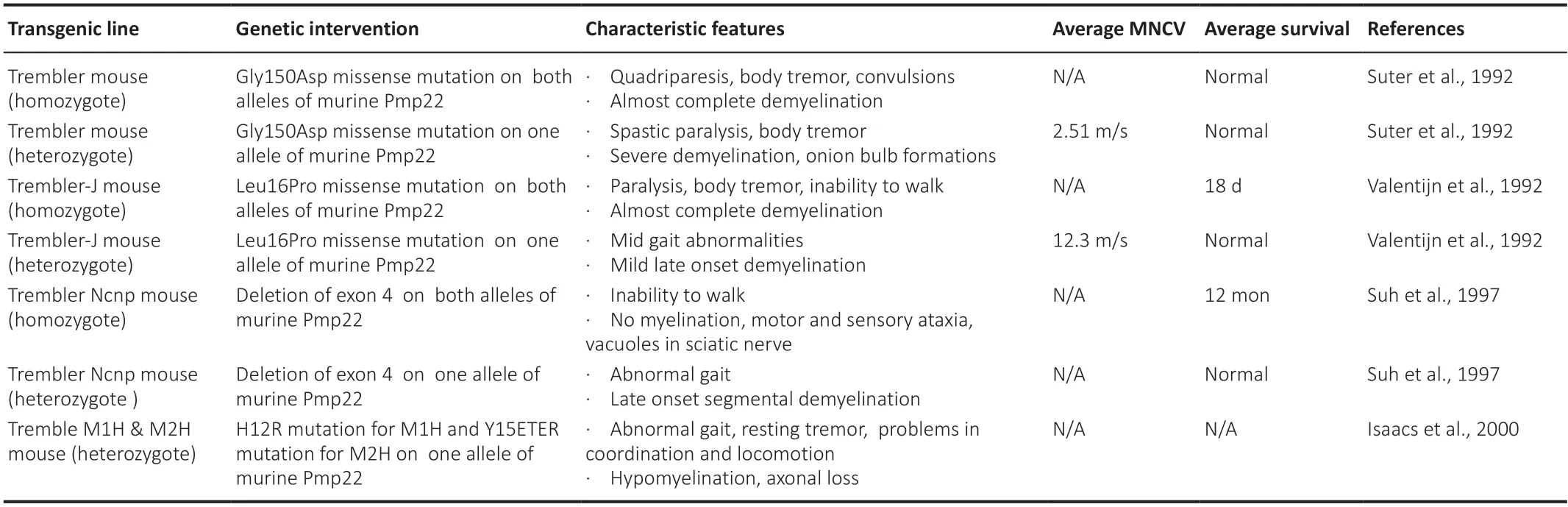
Table 1|CMT1A rodent models: PMP22 mutant models
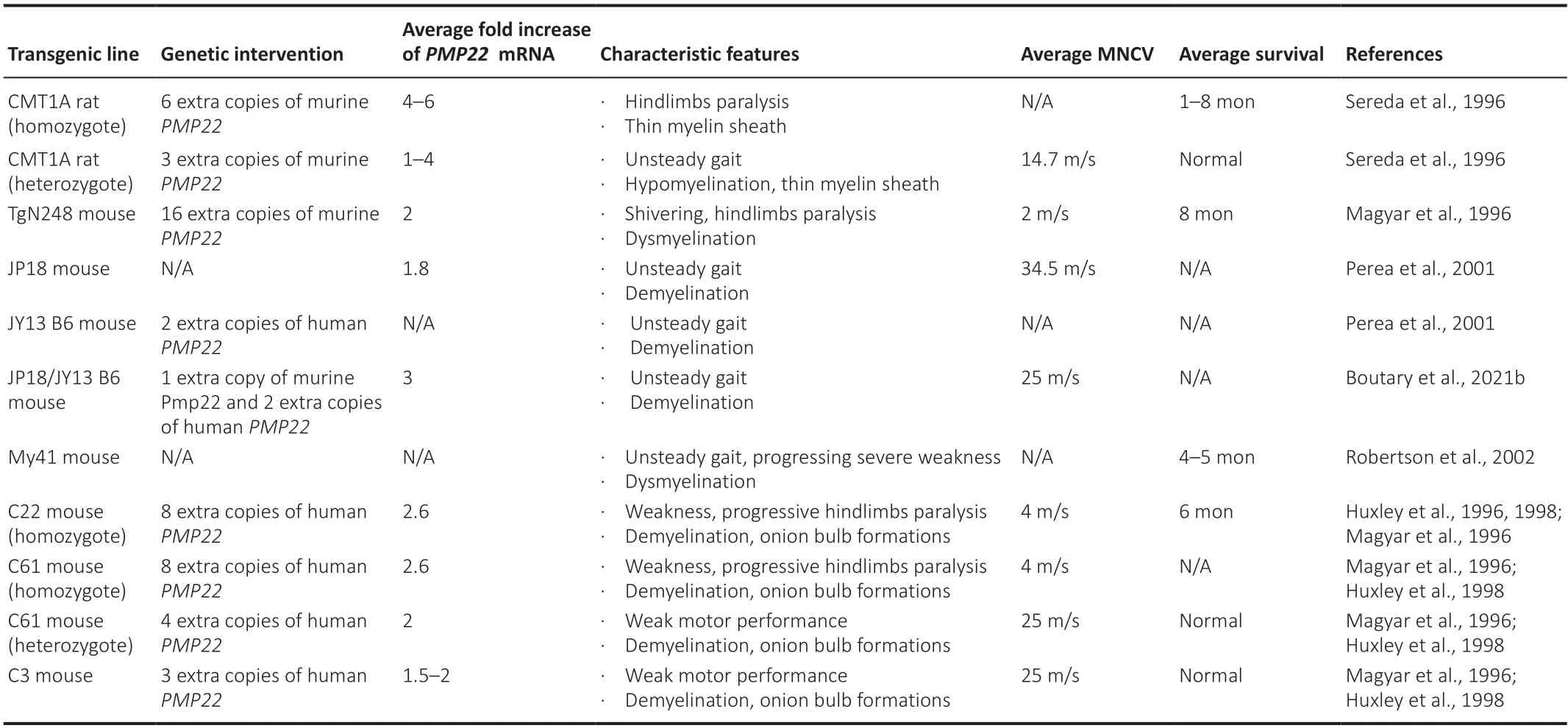
Table 2|CMT1A rodent models: PMP22 overexpression models
To date,there is no effective treatment for CMT1A and its management remains mostly supportive and symptomatic.Therefore,based on the cause of the disease,several efforts have focused on developing gene silencing approaches for CMT1A,all of which are currently at the pre-clinical level(Boutary et al.,2021a;Stavrou et al.,2021b).The overall aim is to reduce the overexpression ofPMP22to normal levels and thereby balancePMP22dosage effects (Boutary et al.,2021a;Stavrou et al.,2021b).SincePMP22has a developmental role in Schwann cell growth and differenti ation,myelogenesis and myelin thickness,it is widely acknowledged that the earlier the therapeutic intervention the more beneficial the treatment will be for CMT1A patients(Stavrou et al.,2022).Although numerous drug therapies have also been proposed to improve the toxic effects ofPMP22overexpression (Stavrou et al.,2021b),with the most clinically advanced being PXT3003 (Attarian et al.,2021;NCT05092841,NCT04762758,NCT03023540,NCT02579759,and NCT01401257),herein we will focus on emergingCMT1Agene therapies.
Search Strategy and Selection Criteria
All years up unti l June 2022 were chosen in our search.These searches were performed in July 2022 using PubMed.Broad search terms such as CMT,CMT1A and gene therapy were used in various combinations.
Gene Therapy Approaches for Charcot-Marie-Tooth Neuropathy Type 1A
Gene therapy describes the delivery of genetic materials into a model or a patient via viral or non-viral vectors.For a successfulCMTgene therapy,it is very important to employ a clinically translatable administration method and thereby achieve the widespread PNS biodistribution of the therapeutic materials (Stavrou et al.,2021b).Unti l today,CMT1A gene therapy approaches have mostly focused on reducing PMP22/PMP22DNA or mRNA (Figure 1).Nevertheless,there have also been approaches trying to ameliorate the CMT1A phenotype by indirect gene therapy methods that are not targetingPMP22.The only CMT1A gene therapies that have proceeded to clinical trials are VM202 (Engensis;NCT05361031),of which no data have been yet shared,and scAAV1.tMCK.NT3 (NCT03520751),which was recently suspended.
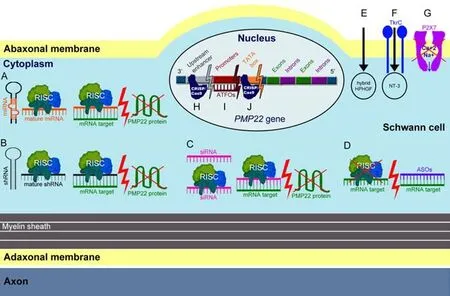
Figure 1|Simplified diagram showing different gene therapy approaches employed to treat CMT1A.
Non-PMP22 directed approaches
Silencing of extracellular ATP-gated ion channel P2X7 purinergic receptor was proposed as an indirect method to ameliorate the CMT1A phenotype (Nobbio et al.,2009).P2X7 receptors are crucial for neuronal synaptic transmission and were shown to have a dosage-depended interaction with PMP22.Hence,it is hypothesized thatPMP22overexpression causes the over-activation of P2X7 receptors,leading to the abnormal influx of Ca2+and Na+,which in turn causes the derangement of Schwann cells (Jarvis and Khakh,2009).Therefore,silencing of P2X7 could be an indirect approach to halt the detrimental cascade of events resulting fromPMP22overexpression.Indeed,in vitrosiRNA,shRNA or pharmacologically mediated silencing of P2X7 in Schwann cell co-cultures,originating from the CMT1A rats,decreased Ca2+influx (Nobbio et al.,2009).Treated cells also presented with elevated levels of myelin-related proteins and ciliary neurotrophic factor (CNTF) release,as well as improved migration properti es (Nobbio et al.,2009).Delivery of the pharmacological antagonist of the P2X7 receptor (A438079) to CMT1A rats improved muscle strength,myelination and motor latencies (Sociali et al.,2016).Long-term administration of P2X7 antagonists has been pre-clinically tested for other diseases without any safety concerns being raised (Dell’Antonio et al.,2002;Honore et al.,2006).However,dose escalation studies of P2X7 antagonists in CMT1A rats suggested that high doses (34 mg/kg) may negatively affect muscle strength and therefore potential administration to humans should follow careful calculation of the therapeutic dose (Sociali et al.,2016).
VM202
VM202 (Engensis;NCT05361031) is a gene therapy approach developed by Helixmith Ltd.(Gangseo-gu Seoul,Korea) based on repeti tive intramuscular injections of a non-viral vector expressing an arti ficial cDNA hybrid of human paracrine hepatocyte growth factor that stimulates PNS regeneration and Schwann cells repairing (Henry et al.,2011;Ko et al.,2018).FDA granted VM202 an orphan drug designation in 2014 and categorized it as a fast track drug in 2016.Subsequently,VM202 has been tested in clinical trials for neurological diseases other than CMT (Sufit et al.,2017;Kessler et al.,2021).However,the beneficial effects of VM202 treatment were progressively attenuated in patients with ischemic heart disease (Kim et al.,2013) and amyotrophic lateral sclerosis (Gordon et al.,2007).Despite these data,Helixmith Ltd.in collaboration with Samsung Medical Center of South Korea launched a VM202 phase I/IIa open-label clinical trial (NCT05361031) for CMT1A patients in 2020 and included 12 participants.This study has been completed;however,no official results have yet been shared.
Neurotrophin-3
Neurotrophin-3 (NT-3) is a crucial neurotrophic factor that activates tyrosine kinase receptor type 3 (Huang et al.,1999),thereby promoting Schwann cell maintenance and regeneration (Patel and Pleasure,2013;Sahenk and Ozes,2020).The potenti al of NT-3 as a therapeutic agent for CMT1A was first assessed using three consecutive subcutaneous injections of 150 µg/kg NT-3 pepti de for 6 months inPMP22mutant Trembler-J mice,immunosuppressed mice hosting xenograft s of CMT1A patients,and eight CMT1A patients (Sahenk et al.,2005).All investigated subjects tolerated NT-3 pepti de administrations well and showed improved axonal regeneration,amelioration of sensory deficits,and better neuropathy scores.To develop a one-off treatment,NT-3 cDNA was then packaged into an AAV1 vector under a muscle specific promoter named triple tandem of muscle creatine kinase (tMCK) (Sahenk et al.,2014;Sahenk and Ozes,2020).Intramuscular administration of 1.5 × 1012vg/kg scAAV1.tMCK.NT3 in Trembler-J mice resulted in therapeutic benefits in the injected and contralateral limbs.In particular,treatment resulted in improved myelin fiber densities,functional and electrophysiological performances for up to 48 weeks post-injection,while it was well tolerated for up to 48 weeks without raising any concerns about toxicity (Sahenk et al.,2014;Sahenk and Ozes,2020).NT-3 gene therapy approach was approved for a phase I/IIa clinical trial (NCT03520751),which was subsequently suspended due to problems with vector production.
CRISPR/Cas9
Clustered regularly interspaced short palindromic repeats/Cas9 (CRISPR/Cas9)approach has been also proposed as a potenti al method to benefit CMT1A patients.In brief,CRISPR is designed to guide Cas9 enzyme toward the DNA or mRNA regions of interest in order to disturb their sequence and either replace,delete or disrupt them.It has been shown thatin vitroCRISPR/Cas9-driven deletion of an upstream super enhancer element of Pmp22 (Pmp22-SE),silencesPmp22mRNA (Pantera et al.,2018).Pmp22-SE is located 90–130 kilobases upstream of thePmp22transcription initi ation site and contains a mark of acetylated methylation of histone H3 on lysine 27 active enhancer;indicating that this element is involved in transcription initiation.It is also speculated that Pmp22-SE is important in tissue specific transcription and in determining if P1 or P2 promoter transcripts are going to be produced(Pantera et al.,2018).It remains to be shown whether this approach could be appliedin vivoand how it may benefit the CMT1A phenotype in models of the disease.
Another CRISPR/Cas9-mediated approach targets the TATA-box of P1 promoter ofPmp22(Lee et al.,2020).In this project,a TATA-targeting CRISPR/Cas9 was non-virally delivered via intraneural injection in C22 mice at postnatal days 6 and 21.After a single injection,mice presented a~40% reduction inPmp22transcript levels that resulted in improved nerve pathology.This was followed by slide improvement in electrophysiological performances that however never approached wild-type levels.Notably,C22 mice,which were used for this research,are a very severe model that does not reflect the phenotype of CMT1A patients.There is a possibility that more robust therapeutic effects could have been observed if a more CMT1A-relevant model,such as C61 het or C3 mice,was employed.Furthermore,for translation purposes,further research should be performed in order to confirm that this method can provide therapeutic benefits in older CMT1A subjects.It is important to acknowledge that this is the onlyPmp22gene silencing approach to confirm no off-target effects by whole genome sequencing.
Anti sense oligonucleoti des
Antisense oligonucleotides (ASOs) are artificial oligomers that complementarily bind on the mRNA sequence of interest and stimulate its degradation.Weekly subcutaneous injection of 25,50 or 100 mg/kg ASOs targeting the open reading frame ofPMP22andPmp22in 5-weekold C22 mice and 6-week-old heterozygote CMT1A rats resulted in dosedependedPMP22/Pmp22silencing as well as in improved myelination and electrophysiological performance of the models for up to 12 weeks postinjection (Zhao et al.,2018).This was the first time to prove that ASOs penetrate the blood-nerve barrier resulting in the total rescue of some neuropathy phenotypes in CMT1A models.These promising results raise the question of whether repeated ASOs-injections can lead to sustained improvement of CMT1A phenotype.In order to address this question it is also important to determine the half-life of the ASOs in the body.
Another ASO-based silencing technology is arti ficial anti parallel triplex-forming oligonucleoti des (ATFOs) that complementarily bind on the targeted exposed region of DNA helix major groove.These molecules inhibit transcription by competing with transcription factors for binding on cis-regulatory regions of the DNA.ATFOs were designed to target purine-rich DNA regions of P1 and P2 promoters ofPMP22,with P2 targeting showing more promising results(Hai et al.,2001).As soon as a suitable purine-rich binding site is identified within the DNA region of interest,ATFOs bind and halt the transcription of the targeted region.What makes ATFOs an appealing gene therapy method is the fact that they can result in robust silencing after binding with a minimal number of DNA molecules while also being responsive to fine tuning of their binding site.However,20 years after their design,none of the sequences has been further tested in a model of CMT1A.
RNA-interference
RNA-interference is a group of short (~22 nucleotides long) nucleotide sequences that complementarily bind on targeted mRNA sequences and either degrade them or block their translation.This group includes small interfering RNAs (siRNA),short hairpin RNAs (shRNA) as well as naturally occurring and artificial microRNAs (miRs).Intraperitoneal injection on postnatal day 6 of a non-viral siRNA that specifically silencesPmp22-L16P mutated allele of Trembler-J mice was shown to improve the CMT1A phenotype of the model (Lee et al.,2017).In detail,Pmp22-targeting siRNA was administrated every third day,five times in total,resulting in improved myelination profile,muscle volume,motor function and electrophysiological performances for up to 3 weeks after the last administration.Similar to the other gene therapy approaches,it would be also relevant to test how this method applies at an older age and in a model that more faithfully reproduces the CMT1A phenotype while also assessing the biodistribution and stability of siRNAsin vivo.
In order to overcome siRNAs degradation by nucleases,another group created a squalenoyl-conjugated siRNA to target a site near the 3′-untranslated region(UTR) ofPMP22(Boutary et al.,2021b).2.5 mg/kg squalenoyl-conjugated siRNA per week were intravenously injected in 16-week-old JP18 and JP18/JY13 mice.This resulted in normalized PMP22 levels leading to a transient improvement of myelination as well as behavioral and electrophysiological performances of the models.Treated animals also showed normalized expression of prior-reduced Krox20,Sox10 (glia markers) and neurofilament(neuronal marker) proteins.
A longer lasting RNA-interference treatment for CMT1A was achieved after packaging an shRNA,targeting the open reading frame of murinePmp22,in an AAV2/9 vector (Gautier et al.,2021).More than 70% of myelinated Schwann cells were transduced after a single intraneural injection with 0.54 vg/dg AAV2/9-shRNA vector into CMT1A rats on postnatal days 6–7.This treatment approach silencedPmp22and improvedMpzexpression,myelination and functionality of the model for up to 12 months postinjection.Since no other PNS tissues were targeted by this treatment,it is sti ll elusive whether this method will be sufficient for treating bigger animals and humans.Another limitation of this approach is that the therapeutic shRNA was designed to target specifically murinePmp22,hence new design and evaluation are needed for the humanPMP22targeting version.Most importantly,what makes intraneural injections less translatable is the risk of fiber damage during injection and the need for concentrated anesthesia during such an invasive procedure (Jeng and Rosenblatt,2011).
Knowing that Schwann cells naturally express miRs that regulate their gene expression,researchers tested the silencing effects of endogenous miRs onPMP22gene.Specifically,on postnatal day 6,C22 mice were intraneurally injected with 7.5 × 104IU/mouse of a lenti viral vector expressing the natural miR318 (Lee et al.,2019).miR318 was shown to target the 3′ UTR ofPMP22gene leading to the silencing ofPMP22/PMP22 mRNA and protein levels in the mouse model of the disease.A single injection with the lenti viral vector expressing miR318 resulted in improved behavioral,electrophysiological and histological readouts for up to 8 weeks later.Along with the limitations of intraneural injection,model employed and age of intervention described above,what makes this approach less appealing for the clinic is the use of an integrating lenti viral vector.
Another naturally occurring miR,miR29a,was shown to bind on a conserved site ofPMP22gene and negatively regulate its translation.Primary cultured Schwann cells originating from C22 mice and human dermal fibroblast cells were treated with synthetic mimics of miR29a or with constructs expressing miR29a,via transient transfection and AAV2-mediated viral transduction.This treatment silencedPMP22mRNA levels,decreased levels of PMP22 protein and restored mitotic acti vity (Serfecz et al.,2019).
More recently,our group developed a novel artificial miR,miR871,which was packaged into an AAV9 vector and was then intrathecally delivered into adult C61 het mice at the translatable concentration of 5 × 1011vg/animal(extrapolating to~2.3 × 1013vg/kg for a 70 kg human) (Stavrou et al.,2022).AAV9-miR871 targets the 3’-UTR of humanPMP22and murinePmp22exon 5.With a single lumbar intrathecal injection,either at the early or late stages of the neuropathy in CMT1A mice,we achieved widespread biodistribution in the PNS leading to a therapeutic benefit for up to 8 months post-injection.In particular,transduction of~50% of Schwann cells by AAV9-miR871 was enough to silencePMP22/Pmp22genes and PMP22/Pmp22 proteins by more than 50% while also enhancing the expression of other myelin related genes and proteins.The effects of AAV9-miR871 were tested in three different treatment groups,which were treated at 2 months of age and analyzed either at 6 or 10 months of age,or treated at 6 months of age and analyzed at 10 months of age.Data showed that silencing effects were followed by improvement in myelination (Figure 2),motor performance,and inflammation of PNS tissues.Furthermore,we demonstrated a reduction in the levels of prior elevated neurofilament light (NF-L) and growth differentiation factor 15 (Gdf15),both circulating biomarkers of axonal degeneration.Early intervention was shown to be more effective as it resulted in the complete reversal of certain CMT1A phenotypes.This indicates that in the early course of CMT1A pathology it is feasible to reverse demyelination and prevent axonal loss.In contrast,secondary axonal loss that occurs at the later stages of the disease is not treatable by restoring Schwann cell function.Nevertheless,all treatment groups are considered to have received post-onset interventions and therefore correspond to the clinically relevant scenario of treating adult CMT1A patients with already progressed demyelination.
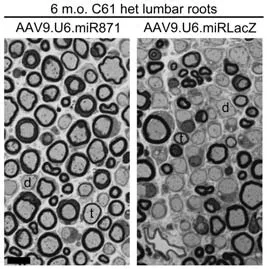
Figure 2|AAV9.U6.miR871-driven silencing of PMP22/Pmp22 genes improves myelination in lumbar roots of C61 het mice.
Critical Summary of the Proposed Charcot-Marie-Tooth Neuropathy Type 1A Gene Therapy Approaches
As described above,numerous CMT1A therapeutic approaches have been proposed and tested at the preclinical level so far.The translation of non-PMP22 directed approaches,such as silencing of P2X7 as well as overexpression of HPHGF or NT-3,is considered rather risky as they may dysregulate secondary pathways leading to a cascade of adverse effects.Moreover,since they do not directly address the disease cause,they are expected to have a lower potential for therapeutic efficacy.They could nevertheless be considered as combination or adjuvant therapies to the more direct approaches.
The CRISPR/Cas9 technology has been employed to silence upstream elements ofPMP22/Pmp22genes (Pantera et al.,2018;Lee et al.,2020).Although CRISPR/Cas9 has already been applied in clinical trials for other diseases,its off-target effects remain a serious concern and a challenge to be addressed (Reardon,2020).In addition,in vivogene editing efficacy in injected tissues remained relatively low at around 11% of Schwann cells (Lee et al.,2020),and further improvement of these rates will be needed to expect therapeutic efficacy.
Subcutaneous administration of ASOs in rodent models of CMT1A has shown silencing of human and murinePMP22/Pmp22genes as well as improved myelination and electrophysiological readouts (Zhao et al.,2018).Although subcutaneous injection is highly translatable and can be even performed by patients themselves,it is debatable whether it will be an efficient delivery route for reaching PNS tissues in humans.Due to the short life of ASOs,toxicity concerns are also raised as it is speculated that repeated injections of high concentrations of ASOs will be required for larger subjects.On the other hand,this short-lived effect of ASOs provides the benefit of allowing easy termination of treatment if any unexpected long-term adverse effects occur.
Intravenous administration of an siRNA encapsulated in squalenoyl nanoparticles targeting humanPMP22gene has shown therapeutic benefit in CMT1A mouse model overexpressing humanPMP22gene (Boutary et al.,2021b).Similar to AAV-mediated miRNA delivery,squalenoylPMP22-targeting siRNA showed significant silencing effects on humanPMP22/PMP22 mRNA and protein,resulting in improved myelination and behavioral performance in disease models.However,these effects resulted after repeated injections twice a week with beneficial effects lasting only for about 3 weeks postinjection.Therefore,further assessment of potential toxic effects after repeated treatments is warranted as well as of the duration of siRNAmediated silencing effects.
Intraneural injections ofPmp22-targeting shRNAs packaged into an AAV2/9 vector silenced Pmp22 protein resulting in improved myelination,behavioral and electrophysiological readouts (Gautier et al.,2021).The effects of this approach on murinePmp22mRNA have not yet been shown.However,regardless of its promising pre-clinical effects,this method has very limited clinical translatability since the shRNA employed is murine-specific and potenti ally contains disruptive mismatches with humanPMP22.In addition,the intraneural injection that has been proposed to deliver the shRNA is not easily translatable,as there is a high risk of fiber damage and anesthesiastimulated toxicity (Jeng and Rosenblatt,2011).These two risks are magnified when considering the limited biodistribution of AAV2/9-shRNA in rodents in accordance with the length of human nerve that would probably require multiple sequenti al injections.
The alternative approach of overexpressing naturally occurring miRs to silencePMP22expression may lead to a cascade of adverse effects on the rest of miRs’ natural targets.On the other hand,AAV9-miR871 was arti ficially designed to target both human and murinePMP22/Pmp22genes and was packaged into the AAV9 vector (Stavrou et al.,2022) that has already been tested and shown to have a good safety profile in humans (NCT03381729,NCT02362438).Furthermore,intrathecal delivery of AAV9-miR871 is considered a non-invasive routine procedure that achieves widespread biodistribution after a single injection not only in mice (Stavrou et al.,2022)but also in bigger animals (Bey et al.,2020).AAV9-miR871 is a one-off treatment that pre-clinically shows long-term beneficial therapeutic effects for at least 8 months post-injection.This approach not only does not stimulate permanent inflammatory reactions in roots,sciatic nerves,dorsal root ganglia and the liver,but it also improves pre-existing inflammation of CMT1A mouse model.Knowing that PMP22 protein is almost exclusively expressed by myelinating Schwann cells (Notterpek et al.,2001;Li et al.,2013),it is expected that ubiquitous expression of any PMP22-targeting silencing molecules would not pose any risk for adverse effects in other cell types andtissues.
It is important to note that,overexpression of miRs,natural or arti ficial,may compete with and dysregulate normal miR-biogenesis leading to adverse effects on transcription regulation and cell signaling.In addition,none of the proposed RNA-interfering approaches (miRNA,shRNA,siRNA,ASOs) have provided extensive off-target analysis.This aspect will be very important to address as such short sequences could potenti ally bind and silence sequences even after incomplete complementarity.
Moreover,it is also fundamentally crucial for the proposed gene therapy approaches for CMT1A to be tested in a model that closely resembles the phenotype of the disease in patients.In the cases when a model with a more severe phenotype is used,it can lead to an invalid underestimation of the therapeutic benefit.Very important is also the age of intervention.When an animal model is treated very early during development and before demyelination and secondary axonal degeneration starts,this can result in overesti mated beneficial effects that will not be reproduced when trying to treat adult CMT1A patients.Finally,all proposed treatments should employ a clinically relevant and safe therapeutic dose along with solid pharmacokinetic and pharmacodynamic data describing the half-life of the therapeutic product within the treated subject.By addressing these parameters,it will make it possible to evaluate the duration of the therapeutic effects and hence it will be feasible to assess the possibility and/or toxicity of repeated dosing.In the cases of virally delivered gene therapies,repeated dosing using the same viral capsid may not be possible due to pre-existing neutralizing anti bodies.
Charcot-Marie-Tooth Neuropathy Biomarkers
Biomarkers are very important readouts that should be able to detect fine changes in disease progression as well as responsiveness to treatment.Currently,there is a high demand for identifying sensitive and clinically relevant biomarkers for developing successful CMT treatments.Until now,most of CMT clinical trials were employing different CMT neuropathy scores in order to evaluate therapeutic responses in patients.However,these outcome evaluation tools showed low sensitivity in detecting treatment responses,particularly in CMT1A patients with a slowly progressive phenotype.Therefore,in recent years some more sensitive biomarkers have emerged that could complement clinical evaluation tools for developing a more comprehensive evaluation of treatment outcomes for CMT neuropathies.
Imaging and tissue biomarkers
CMT progression is directly associated with muscle atrophy and fat accumulation.These aspects can be evaluated using magnetic resonance imaging (MRI) for measuring fat accumulation in lower limb muscles (Morrow et al.,2018;Bas et al.,2020) or in the endoneurial space of the sciatic nerve (Kim et al.,2021).In addition,diffusion tensor imaging for peripheral nerve integrity (Cheah et al.,2021) and quantitative echogenicity with neuromuscular ultrasound (Kitaoji et al.,2021) are also considered sensitive CMT progression readouts.
Recently,intraepidermal nerve fiber density was proposed to relate with disease severity as skin punch biopsies of CMT1A patients showed reduced numbers of Merkel cells,high levels of denervated Merkel cells,shortened paranodes and decreased fraction of long nodes (Hartmannsberger et al.,2020).Therefore,intraepidermal nerve fiber density could be considered as a supplementary CMT1A biomarker that could potenti ally be applied to other CMT types too.An animal study also proposed gait parameters as a CMT1A biomarker,however still remains to clarify if this readout will be sensitive enough for patients (Hwang et al.,2021).
Dermal skin biopsies were also used to quantifyPMP22mRNA levels of myelinated nerve fibers using nanostring RNA analysis (Fledrich et al.,2012;Svaren et al.,2019).Since most of the proposed CMT1A treatments aim at reducing overexpressedPMP22levels,it is of great value to have a noninvasive and reliable biomarker measuringPMP22levels in the clinic.
Circulating biomarkers
Blood biomarkers are a rapidly emerging tool that has great translatability potenti al.In the blood stream there are circulating markers that correspond to CMT severity by reflecting Schwann cell abnormalities,axonal degeneration and muscular denervation.
Plasma NF-L level is a reliable circulating biomarker,the elevation of which is proportional to axonal degeneration (Sandelius et al.,2018;Millere et al.,2021;Rossor et al.,2021;Amaador et al.,2022;Rossor and Reilly,2022).CMT patients (Sandelius et al.,2018;Millere et al.,2021;Rossor et al.,2021)and a CMT1A animal model (Stavrou et al.,2022) have shown increased levels of NF-L.In CMT patients,NF-L levels are also correlated with disease severity,as measured by CMTES and CMTNS functionality scores,as well as with CMT patients’ age (Sandelius et al.,2018).Importantly,treated CMT1A animal models have shown decreased levels of NF-L,indicating that this is a treatment-responsive biomarker (Stavrou et al.,2022).Whether NF-L levels in patients would also be treatment responsive remains to be determined.
Transmembrane protease serine 5 (TMPRSS5;also known as spinesin) is highly expressed in Schwann cells where it cleaves trypsin (Yamaguchi et al.,2002;Watanabe et al.,2004;Wang et al.,2020).TMPRSS5 is also an informative blood biomarker as it was shown to be chronically upregulated during CMT1A progression (Wang et al.,2020).However,in contrast to NF-L,TMPRSS5 did not correlate with any of the CMT scoring systems,electrophysiological readouts,or with age.This makes TMPRSS5 a less useful tool for reflecting treatment responsiveness.Further studies are needed to also clarify whether TMPRSS5 is a conserved and treatment-responsive biomarker in CMT animal models.
Proteomics analysis recently revealed novel CMT blood biomarkers,such as neural cell adhesion molecule-1 (NCAM1) and Gdf15 (Jennings et al.,2022).NCAM1 regulates synapse rearrangement and regeneration while Gdf15 levels are analogous to axonal degeneration.Similar to TMPRSS5,NCAM1 and Gdf15 levels have only been validated as biomarkers that distinguish CMT patients from healthy individuals,but they have not yet been correlated with CMT outcome measures.Similar to CMT1A patients,C61 het animals showed elevated levels of Gdf15 at progressed disease stages as well as treatment responsiveness of serum Gdf15 following gene therapy (Stavrou et al.,2022).Although no NCAM1 data have yet been published for CMT1A animals,a CMT1X animal model showed elevated NCAM1 levels in advanced disease stages that were reversed after gene therapy treatment (Kagiava et al.,2021).
Circulating microRNAs (miRs) are also promising biomarkers.Schwann cell and muscle derived miRs are proposed to reflect myelination status and muscle degeneration,respectively (Wang et al.,2021).Circulating myomiR206 and myomiR133a were found elevated in CMT1A patients,with the latter showing a strong correlation with NF-L levels (Wang et al.,2021).MyomiR133a and myomiR1 showed a negative correlation with CMAP scores but a positive correlation with CMT neuropathy scores (Wang et al.,2021).The levels of all the above myomiRs correlated with age.Circulating Schwann cell derived miR328 and miR223-3p correlated with TMPRSS5 and NF-L levels as well as with CMT neuropathy scores but not with age (Wang et al.,2021).Similar to previous biomarkers circulating miRs have not yet been tested for their treatment responsiveness and conservation among different species.
Conclusion
CMT1A gene therapy is a rapidly emerging and promising field.We suggest that direct silencing of overexpressedPMP22closer to physiological levels is the most promising and effective approach to reverse and restore the genetic mechanism of CMT1A.Additional indirect approaches,including some drugbased approaches not discussed in this review,may offer added benefits in a combination treatment.Unti l today,most proposed CMT1A gene therapies remain at a pre-clinical stage of development.However,extensive natural history data,lessons learned from previous drug treatment trials in CMT1A patients (Boutary et al.,2021a;Stavrou et al.,2021b),as well as the discovery of treatment-responsive CMT biomarkers,are expected to facilitate the optimization of CMT1A clinical trial design.
Many challenges remain to be addressed before translating a CMT1A gene therapy approach to the clinic.Given that this is not a life-threatening disease,potenti al side effects should be carefully predicted and excluded.Hence,we propose confirming the proof-of-concept of the suggested therapy in more than one animal model when available,using more than one therapeutic dose in a dose-escalation study,while also performing extensive toxicity studies in rodents as well as in larger animals.
Author contributions:MS and KAK conceived,wrote and edited themanuscript.MS generated Tables and Figures.KAK reviewed the manuscript.Both authors approved the final version of the manuscript.
Conflicts of interest:The authors declare that there is no conflict of interest regarding the publication of this paper.
Open access statement:This is an open access journal,andarticles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Bystanders or not? Microglia and lymphocytes in aging and stroke
- Alzheimer’s disease risk after COVID-19: a view from the perspective of the infecti ous hypothesis of neurodegeneration
- Serine and arginine rich splicing factor 1: a potenti al target for neuroprotection and other diseases
- Can glial cells save neurons in epilepsy?
- Lights for epilepsy: can photobiomodulation reduce seizures and offer neuroprotection?
- The landscape of cognitive impairment in superoxide dismutase 1-amyotrophic lateral sclerosis