
Overcoming mitochondrial dysfunction in neurodegenerative diseases
2023-02-24 05:24JoPessoaAnaDuarte
中国神经再生研究(英文版) 2023年7期
João Pessoa,Ana I.Duarte
Due to their intense electrical activity,neurons have high energy demands.This requirement makes them particularly sensitive to mitochondrial dysfunction.Like all eukaryotic cells,neurons have intrinsic mechanisms to mitigate the impact of mitochondrial dysfunction and its consequent production of toxic substances.Among such(neuro)protective mechanisms,mitochondrial autophagy (mitophagy) is responsible for the removal of dysfunctional mitochondria.Pathological inhibition of mitophagy,together with insufficient mitochondrial activity,results in a shortage of adenosine triphosphate (ATP)and the accumulation of reactive oxygen species(ROS) (Simmons et al.,2020).These alterations may trigger extensive apoptotic neuronal death(Figure 1A) which,together with the post-mitotic nature of neurons,impedes the replacement of the apoptotic cells.This irreversible loss of neurons may underlie the progressive decline in the function of the central nervous system,culminating in the arousal of neurodegenerative diseases (Simmons et al.,2020).The actual role of mitochondrial dysfunction has been increasingly demonstrated in Alzheimer’s disease.For example,post-mortem analysis of patient brains has revealed the reduced expression and acti vity of mitochondrial respiratory chain complexes(Troutwine et al.,2022).
Although the causing mechanisms of neurodegenerative diseases are diverse and still poorly characterized,mitochondrial dysfunction may be a common driver of major neurodegenerative diseases (Ebanks and Chakrabarti,2022).As such,overcoming neuronal mitochondrial dysfunction provides a therapeutic opportunity to target a common problem in multiple neurodegenerative diseases.Among the most promising therapeutic strategies,we emphasize those involving the replacement of dysfunctional mitochondria (Simmons et al.,2020) or the enhancement of ATP production pathways in the cytoplasm (Cai et al.,2019;Grieco et al.,2019;Zhang et al.,2021).However,a major challenge to the development of efficient pharmacologic interventions in the brain has been the reduced permeability of the blood-brain barrier to most therapeutic molecules (Abbott et al.,2010).
From the above,we present an overview of the most recent promising approaches to overcome neuronal mitochondrial impairment,and of the new strategies to deliver mitochondriotropic drugs into the brain,to treat neurodegenerative diseases.
Increasing mitochondrial fitness:Within mitochondria,ATP synthase is not only the major ATP producer,but it also constitutes an important target for oxidative modifications mediated by ROS upon several neurogenerative diseases (namely Alzheimer’s disease).Thus,drugs that protect this protein complex from oxidative modifications may preserve its function and ultimately protect neurons from degeneration (Ebanks and Chakrabarti,2022).However,since mitochondrial oxidative damage is not restricted to ATP synthase,but may instead affect every mitochondrial component,it is plausible that the replacement of dysfunctional mitochondria may also constitute a promising neuroprotective strategy (Simmons et al.,2020).Such a mitochondrial replacement can be achieved through the induction of mitochondrial biogenesis,e.g.,through the conjugation of the transcriptional activation in the nuclear genome,replication of mitochondrial DNA,and biosynthesis of lipid membranes and mitochondrial proteins(Simmons et al.,2020).In this respect,the peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) is frequently regarded as the most important regulator of the expression of nuclear genes involved in mitochondrial biogenesis.This protein is known to recruit several transcription factors that activate the transcription of nuclear genes coding for essenti al proteins of mitochondrial biogenesis.Importantly,preclinical studies indicate that the induction of mitochondrial biogenesis may constitute a promising therapeutic strategy against diseases of the central nervous system (Simmons et al.,2020).An alternative approach to the induction of mitochondrial biogenesis is mitochondrial augmentation therapy,which includes the collection of hematopoietic stem and progenitor cells from individuals with diseases caused by genetic defects in mitochondrial proteins.Then,these cells areex vivoenriched with mitochondria from healthy donors and returned to the patient’s bloodstream to improve mitochondrial function(https://minoviatx.com/therapy/),and eventually counteract neurodegenerative diseases.An increase in healthy mitochondrial content in the cell may also require an increased supply of oxygen and sugar.Since the dysregulation of the blood flow to the brain constitutes a prominent feature of Alzheimer’s disease patients (Zhu et al.,2022),such an increased blood irrigation elicited by the “new” mitochondria would be accompanied by an increased metabolic demand of the brain tissues and further enhanced by vascularization factors.Nevertheless,we must emphasize that mitochondrial function is generally exacerbated in tumors and,therefore,we cannot exclude the risk for these mitochondrial interventions to promote cancer progression.As such,it should be executed only in cells under neurodegeneration (Simmons et al.,2020).
Another important limitation of mitochondrial biogenesis induction and mitochondrial augmentation therapy is their failure to remove damaged mitochondria,which frequently carry mutations or deletions in their DNA.Computer simulations have shown that deletions in mitochondrial DNA are likely to facilitate the replication of these shorter DNA molecules(Holt and Davies,2021).Consequently,the increased propagation rates of genetically aberrant mitochondria will likely exacerbate their defective bioenergetic function.The mitochondrial genome,notably,in its copy number,also affects the bioenergetic capacity of these organelles.Important determinants of the mitochondrial DNA copy number are the DNA polymerase gamma,which catalyzes its replication,and the mitochondrial transcription factor A,which regulates both its transcription and compaction,protecting it from oxidative chemical modifications.Increased expression levels of these two molecular players have been related to increased copy numbers of the mitochondrial genome and increased bioenergetic capacity of mitochondria (Filograna et al.,2021).As such,their modulation could also be a promising approach to increasing mitochondrial fitness.
Mitochondria are also the major cellular sources of oxidative stress,which easily results in the oxidation of mitochondrial DNA by ROS.One of its major oxidation products,8-oxoguanine,can induce the proinflammatory release of mitochondrial DNA into the cytoplasm (Riley and Tait,2020).The major mitochondrial DNA repair enzyme,8-oxoguanine DNA glycosylase,removes 8-oxoguanine from the mitochondrial genome.The activity of this enzyme could be enhanced with small molecule activators to protect mitochondrial DNA integrity and attenuate mitochondrial dysfunction (Tian et al.,2022).Although most mitochondrial proteins are nuclear-encoded,the correct functioning of the mitochondrial respiratory chain is dependent on several mitochondrial-encoded proteins.As such,the protection of the mitochondrial genome from oxidative damage could hold great promise in overcoming mitochondrial dysfunction.Another protector against ROS-mediated molecular damage is the transcription factor NF-E2 p45-related factor 2 signaling pathway.Despite some issues in their target specificity,effectiveness,and safety,activators of this pathway could provide a protective strategy against a vast array of diseases,including neurodegenerative ones (Cuadrado et al.,2019).
In the absence of repair,mitophagy provides the most effective process of removing mitochondria with mutations in their DNA.This goal has been pursued through strategies to increase the acti vity of the PTEN-induced kinase 1,a potent mitophagy inducer,whose stimulation could prevent inflammation,apoptosis,and neuronal cell death(https://www.mitokinin.com/our-science/pink1/).The regulation of mitophagy and apoptosis are also affected by the mitochondrial permeability transition pore (Bonora et al.,2022).Inhibition of this translocation complex with small molecules has been tested for the treatment of Parkinson’s and other neurodegenerative diseases through the inhibition of neuroinflammation and prevention of neuronal cell death (http://www.nrgtherapeutics.com/).
Increasing cytoplasmic glucose metabolism:Additional therapeutic opportunities reside in the cytoplasm,whereby the neuroprotective effects have been related to the enhancement in cellular uptake of glucose and its cytoplasmic degradation(Griffith et al.,2018;Cai et al.,2019;Grieco et al.,2019;Zhang et al.,2021).
Altered insulin levels in the brain,either defective or excessive,have been associated with an increased risk for Alzheimer’s disease (Griffith et al.,2018).As in other cell types,insulin induces the translocation of glucose transporters to the neuronal cell membrane,increasing their cytoplasmic levels of glucose (Griffith et al.,2018).Neurons from Alzheimer’s disease patients have shown hyperglycemia and decreased expression of glucose transporters (Zhang et al.,2021),and the maintenance of neuronal glucose homeostasis depends on adequate insulin levels.Treatments that fine-tune insulin signaling resulted in improved cognitive function,both in healthy individuals and Alzheimer’s disease patients (Griffith et al.,2018).Insulin release can be induced by glucagon-like peptide 1 (GLP-1),a gut-produced hormone.Studies in non-diabetes animal models have shown that GLP-1 receptor agonists (GLP-1RAs) could have neuroprotective effects against Alzheimer’s and Parkinson’s diseases,and others.These effects were likely mediated by the inhibition of oxidative stress,inflammation,and apoptosis (Grieco et al.,2019).These observations demonstrate that hormone signaling interventions that restore glucose uptake into the brain are promising therapeutic approaches against neurodegeneration.
In addition to glucose uptake,glycolysis is also impaired in several neurodegenerative diseases,including Alzheimer’s,Parkinson’s,Huntington’s diseases,and amyotrophic lateral sclerosis (Zhang et al.,2021).This finding suggests enhancing glycolysis as a potenti al neuroprotective approach(Zhang et al.,2021).Accordingly,terazosin,an activator of phosphoglycerate kinase 1,the first glycolytic enzyme directly producing ATP,could slow down neurodegeneration in Parkinson’s disease mice (Cai et al.,2019).Terazosin also decreased the progression of motor disability in Parkinson’s disease patients (Cai et al.,2019).These results suggest that terazosin may attenuate Parkinson’s disease by promoting glycolysis.
Although glucose is the preferred energy source of the brain,ketone bodies provide an important alternative energy substrate.Despite the lack of compelling clinical evidence,diet supplementation with medium-chain fatty acids,which generates the β-hydroxybutyrate,could be neuroprotective.This ketone body has been proposed to inhibit mitochondrial failure,oxidative stress,and neuroinflammation (Jensen et al.,2020).
Energy conversion in cells is deeply associated with the consumption of oxygen.Depending on its severity,hypoxia (decreased oxygen availability caused by its low supply or increased consumption) can be either neurotoxic or neuroprotective (Han et al.,2021).Humans living at high altitudes had decreased prevalence of neurodegenerative diseases (Han et al.,2021).These findings encourage the development of neuroprotective strategies that mimic hypoxia,namely those involving the hypoxia-inducible factor 1α (HIF-1α) transcription factor.Under sufficient cellular oxygen levels,HIF-1α is targeted for degradation.Under hypoxia,it is stabilized and imported into the nucleus,where it activates the transcription of genes that promote glycolysis and decrease mitochondrial respiration (Han et al.,2021).Thus,therapeutic overexpression of HIF-1α,which up-regulates glycolysis,could be an intervention against neurodegeneration.
Increasing glucose uptake and its cytoplasmic metabolism not only alleviates energy deficiency but may also counteract excessive ROS production(Tang,2020).Enhanced glucose uptake upregulates the pentose phosphate pathway and consequently increases NADPH production and the levels of reduced glutathione,a pivotal anti oxidant defense (Tang,2020).Besides the critical role in bioenergetics,increased ATP levels may also promote the dissolution of protein aggregates often observed in patients with neurodegenerative diseases (Tang,2020).
Importantly,increasing the cytoplasmic metabolism of glucose without the concomitant recovery of mitochondrial function may be little effective,or even risky.This is because the alterations in the electron flow of the mitochondrial respiratory chain can promote ROS production,which can further damage mitochondrial DNA and other cellular components(Riley and Tait,2020).In addition,the electron donors generated in glycolysis would be fruitless if not usable by mitochondria.Moreover,the energetic yield of glycolysis (2 molecules of ATP per molecule of glucose) is well below that of Krebs’ cycle and respiratory chain (36 molecules of ATP per molecule of glucose).For all these reasons,the increase in cytoplasmic glucose metabolism should complement,and not replace,the increase in mitochondrial fitness.
Altogether,these studies indicate the upregulation of ATP-producing metabolic pathways in the cytoplasm as a promising therapeutic approach to neurodegenerative diseases.Increasing glucose uptake is a necessary step to up-regulate its cytoplasmic degradation.The latter seems to compensate for decreased ATP production in mitochondria.The mechanisms that cause neurodegenerative diseases are complex.Nevertheless,the findings described above demonstrate that several metabolic pathways can be targeted to overcome mitochondrial dysfunction,providing multiple therapeutic opportunities against neurodegeneration.
Delivering drugs into the brain:A major challenge in the pharmacologic treatment of neurodegenerative diseases is the achievement of efficient drug delivery into the brain.Brain delivery requires that an intravenous drug crosses the blood-brain barrier,which is formed by the endothelial cells of the brain capillary vessels.To protect the brain from toxic substances,the blood-brain barrier contains tight junctions between adjacent cells that ensure a remarkably low permeability to blood molecules (Abbott et al.,2010).Consequently,this essenti al protective barrier complicates intravenous drugs from reaching the brain.This limitation has been frequently overlooked in drug research for neurodegenerative diseases (Pardridge,2019).In addition,the P-glycoprotein,an active efflux system of the brain capillary vessels,pumps small molecules out of its endothelial cells,further complicating their crossing of the blood-brain barrier (Pardridge,2019).Thus,drugs that are P-glycoprotein substrates will face an additional obstacle to their brain delivery.
Drug injection into the cerebrospinal fluid,which surrounds the surface of the brain,allows the drug to reach its surface,but not to achieve a homogeneous distribution throughout this organ(Pardridge,2019).This goal has been pursued through the exploitation of transport systems of the endothelial cells in the blood-brain barrier.Small molecules failing to diffuse through the membranes of these cells can be engineered to interact with small-molecule transporters on those membranes,including the GLUT1 glucose transporter (Pardridge,2019;Figure 1B,left).
Endothelial cells of the blood-brain barrier also contain transporters for peptides and proteins,including the insulin receptor (Abbott et al.,2010).They work by forming a vesicle around the protein or pepti de and translocating it across the endothelial cell.A therapeutic protein or pepti de can be fused to a monoclonal anti body that mimics a substrate of one of these transporters,yielding a molecular Trojan horse (Pardridge,2019).Alternatively,a gene coding for the therapeutic protein or pepti de can be provided through gene therapy.The gene can be packed inside adenoassociated 9 viruses and injected into the blood(Gosselet et al.,2021).These viruses are generally safe but poorly translocated through the bloodbrain barrier.Thus,non-viral alternatives for gene delivery exist and include cationic lipid micelles and encapsulation inside liposomes or lipid nanoparticles,which are coated (via polyethylene glycol molecules) with monoclonal antibodies that interact with blood-brain barrier receptors(molecular Trojan horses;Figure 1B,right).Offtarget effects are minimized through the uti lization of tissue-specific gene promoters (Pardridge,2019).In comparison with adeno-associated 9 viruses,this approach can accommodate larger DNA molecules and has shown higher efficiency of gene delivery into the brain after intravenous injection.Its conjunction with transient receptor opening,which can be induced chemically or with ultrasounds,seems to be the most promising approach (Gosselet et al.,2021).
Concluding remarks:The recent development of strategies to deliver drugs into the brain opens exciting perspectives in the treatment of neurodegenerative diseases.Although these promising technologies have been mostly exploited to target specific mechanisms of Alzheimer’s and Parkinson’s diseases (Pardridge,2019),they could also successfully target a common player in the major neurodegenerative diseases: mitochondrial dysfunction.
Mitochondrial dysfunction can be mitigated through the induction of its biogenesis.It can also be bypassed through the increase in glucose uptake and glycolysis.These processes can be promoted through the neuronal uptake of specific small molecules that can activate endogenous genes or directly up-regulate specific pathways(e.g.,terazosin,an activator of the first ATPproducing glycolytic enzyme;Figure 1C).These small molecules should be carefully engineered to mimic substrates of blood-brain barrier transporters.As an alternative to transcriptional activation from endogenous genes,neurons could be supplied with exogenous genes,whose expression will increase the levels of GLP-1RAs,HIF-1α,and PGC-1α.GLP-1RAs will promote glucose uptake,increasing its availability to glycolysis,which in turn can be up-regulated by HIF-1α (Figure 1C).PGC-1α induces mitochondrial biogenesis (Figure 1C),which further increases ATP production.
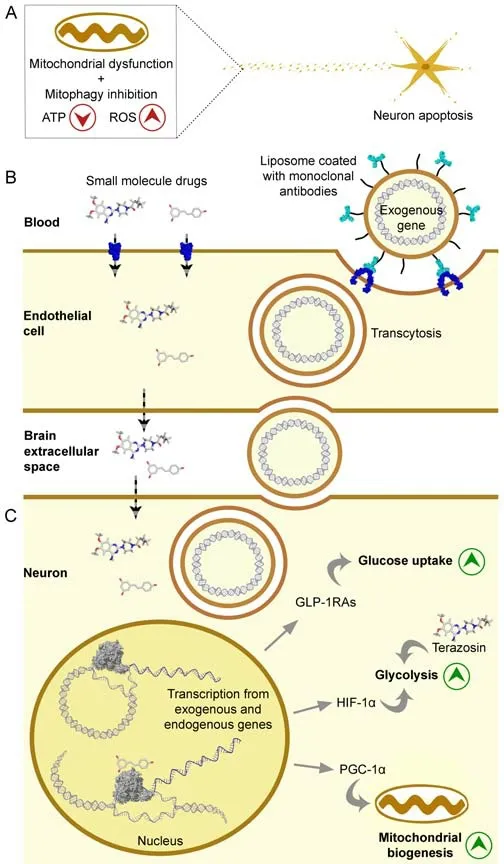
Figure 1|Strategies to overcome mitochondrial dysfunction in neurodegenerative diseases.
Important challenges and limitations of these approaches include the achievement of brain specificity and precise fine-tuning of protein levels.The latter could potentially be attained through exogenous gene expression from inducible promoters,using an inducer able to cross the blood-brain barrier.Importantly,scaling up the production of encapsulated transgenes appears to be a major challenge (Pardridge,2019).Moreover,the elevated costs of a drug tend to negatively influence the decision of their prescription to geriatric patients.Therefore,additional efforts are needed to lower the costs of these therapies.
In conclusion,there are several promising strategies to overcome mitochondrial dysfunction,as well as to deliver drugs into the brain.The intersection of these two endeavors is a timely opportunity to tackle neuronal mitochondrial dysfunction,a common driver of neurodegenerative diseases.
This work was financed by the European Regional Development Fund (ERDF),through the COMPETE 2020– Operational Programme for Competi tiveness and Internationalization,under the project POCI-01-0145-FEDER-029391(Mito4ALS);by Portuguese national funds via FCT–Fundação para a Ciência e a Tecnologia,under the projects UIDB/04539/2020,UIDP/04539/2020,LA/P/0058/2020,and UIDB/00081/2020;and by the European Social Fund,through the DL57/2016 -SFRH/BPD/84473/2012 to AID.
João Pessoa*,Ana I.Duarte
CNC– Center for Neuroscience and Cell Biology,CIBB– Center for Innovative Biomedicine and Biotechnology,University of Coimbra,Coimbra,Portugal
*Correspondence to:João Pessoa,PhD,joao.pessoa@cnc.uc.pt.
https://orcid.org/0000-0002-9202-5728(João Pessoa)
Date of submission:July 5,2022
Date of decision:September 23,2022
Date of acceptance:October 20,2022
Date of web publication:November 9,2022
https://doi.org/10.4103/1673-5374.360279
How to cite this article:Pessoa J,Duarte AI(2023) Overcoming mitochondrial dysfunction in neurodegenerative diseases.Neural Regen Res 18(7):1486-1488.
Open access statement:This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Bystanders or not? Microglia and lymphocytes in aging and stroke
- Alzheimer’s disease risk after COVID-19: a view from the perspective of the infecti ous hypothesis of neurodegeneration
- Serine and arginine rich splicing factor 1: a potenti al target for neuroprotection and other diseases
- Can glial cells save neurons in epilepsy?
- Lights for epilepsy: can photobiomodulation reduce seizures and offer neuroprotection?
- The landscape of cognitive impairment in superoxide dismutase 1-amyotrophic lateral sclerosis