
Transforming cancer cells for long-term living with cancer:An inspiring new approach
2023-05-14 03:44MingjieJiangDiannaGuFuraoLiuChenjingLinLingTian
Mingjie Jiang ,Dianna Gu ,Furao Liu ,Chenjing Lin ,Ling Tian
1Department of Head and Neck Surgery,Sun Yat-sen University Cancer Center,State Key Laboratory of Oncology in South China,Collaborative Innovation Center for Cancer Medicine,Guangzhou 510060,China;2 Department of Chemotherapy,the First Affiliated Hospital of Wenzhou Medical University,Wenzhou 325000,China;3Shanghai Key Laboratory of Pancreatic Diseases,Shanghai General Hospital,Shanghai Jiao Tong University School of Medicine,Shanghai 201620,China;4 Department of Central Laboratory,Shanghai Chest Hospital,Shanghai Jiao Tong University School of Medicine,Shanghai 200030,China
Abstract Cancer is the leading cause of human death and imposes a huge health burden.Currently,no matter what advanced therapeutic modalities or technologies are applied,it is still peculiarly rare for most cancers to be radically cured whereas therapy resistance and tumor recurrence are ever so common.The long-standing cytotoxic therapy is hard to achieve long-term tumor control,and produces side-effects or even promotes cancer progression.With growing understandings of tumor biology,we came to realize that it is possible to transform but not kill cancer cells to achieve long-term living with cancer,and directly altering cancer cells is a promising way.Remarkably,tissue microenvironment is involved in the fate determination of cancer cells.Of note,leveraging cell competition to combat malignant or therapy-resistant cells shows some therapeutic potentials.Furthermore,modulating tumor microenvironment to restore a normal state might help to transform cancer cells.Especially,reprogramming cancer-associated fibroblasts,and tumor-associated macrophages,or normalization of tumor vessel,tumor immune microenvironment,and tumor extracellular matrix or their combinations,et al.,revealed some long-term therapeutic benefits.Despite the massive challenges ahead,it would be possible to transform cancer cells for longterm cancer control and living with cancer longevously.The related basic researches and corresponding therapeutic strategies are also ongoing.
Keywords: Cancer treatment;cancer biology;cancer microenvironment;microenvironment normalization;cell competition;cell reprogramming
Introduction
Cancer has become the leading cause of human death worldwide.In recent decades,a huge number of studies revealed that accumulated gene mutations and/or chromosomal abnormalities in tumor cells drive tumor initiation and progression.Thus,it became the primary strategies of tumor control to kill tumor cells due to the irreversibility of genetic alterations (1).More importantly,the clinical evaluation of therapy efficacy is largely dependent on the effect of tumor cell killing,such as the Response Evaluation Criteria in Solid Tumors (RECIST).Actually,cytotoxic therapy,including chemotherapy,radiotherapy,and targeted therapy,etc.,firmly holds the mainstream regimens for current cancer treatment.Besides,the maximum tolerable dose (MTD) of the cytotoxic drugs is usually applied,which means that as long as the normal tissue can withstand,the dose will be steadily scaled up for the maximum tumoricidal efficacy.
Although remarkable curative effects of cytotoxic therapy have been achieved to a sprinkling of tumors,such as trophoblastic tumor,acute lymphocytic leukemia (ALL),etc.,the therapeutic effects of most cancer treatments based on tumor cell killing remain unsatisfactory (2).Some efforts to improve the tumoricidal effects are ongoing,such as developing new drugs or rescheduling the dose and fractions.However,the therapeutic modalities of killing cancerous cells have inevitably encountered bottlenecks(2,3).Fortunately,with a growing understanding of tumor biology,especially with respect to tumor microenvironment (TME),it becomes possible to develop new strategies other than directly killing tumorous cells.In particular,we come to realize that the malignant behavior of cancer cells is not inherently unchanged,and cancer cells can be educated to become less aggressive (4-8).Hence,it seems to be more imperative to revisit the biology of cancer and re-examine current cancer treatment strategies,which would contribute to accordingly developing long-term control strategies for cancer management.
Dilemmas of current cancer cytotoxic therapy
Unfortunately,despite oncologists have spared no efforts to develop and improve cytotoxic regimens for more than 50 years,at least several dilemmas in cytotoxic therapy remain insurmountable in the war on cancer (Figure 1).
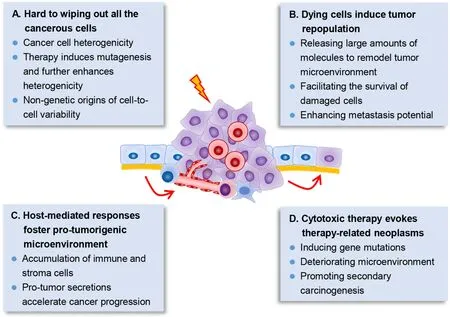
Figure 1 Several dilemmas in cancer cytotoxic therapy.(A) Cancer cells show significant heterogenicity,both at the genetic and epigenetic level and even nongenetic variations.Therapy further enhances mutagenesis which may promote heterogenicity;(B) Therapy-induced dying tumor cells can release large amounts of molecules,which modulate the tumor microenvironment and further promote survival,proliferation and metastasis of the surviving tumor cells;(C) Cytotoxic therapy also induces damages in host normal cells,which further recruit immune and stroma cells via their secretions,and contribute to promoting cancer progression;(D) Cytotoxic therapy further induces secondary carcinogenesis.Dying tumor cells’ secretions,therapy-induced gene mutations and deteriorated microenvironment may conduce to promoting secondary carcinogenesis.
Wiping out all cancerous cells is hard to accomplish
To preserve the architecture and function of normal tissues,the dose of anti-tumor drugs could not be scaled up indefinitely.More crucially,cancer cells within the tumor tissues exhibit significant heterogeneity at both genetic and transcriptional level,even that every single cancer cell within the tumor is different (9).Cell fusion further elevated the level of phenotypic plasticity and might substantially accelerate tumor adaptation to new therapeutic pressures (10).In that,there are large odds that therapy resistant cells have already existed in the tumor,and that the adopted therapy just kills the sensitive cells while the resistant cells can survive and cause tumor recurrence (11).
What’s more,chemoradiation significantly boosts DNA mutations.Cancer cells under specific targeted therapy down-regulated mismatch repair (MMR) and homologous recombination DNA-repair genes,and concomitantly upregulated error-prone polymerases to induce higher mutation (12).The acquired resistance of epidermal growth factor receptor (EGFR)-mutant non-small-cell lung cancers (NSCLC) to EGFR inhibitors could be either selected from pre-existing EGFR(T790M)-positive clones or evolved from initially EGFR(T790M)-negative drugtolerant cells (13).Moreover,mutated KRAS (KRAS proto-oncogene,GTPase) clones that evolved from colorectal cancer patients during EGFR inhibition treatment declined upon withdrawal of therapy (14),indicating that therapy plays an important role in sustaining therapy-resistant genotype.In addition,epigenetic mechanisms also actively participate in promoting therapy resistance,and cause reversible drugtolerant state in cancer cell subpopulations (15).Nongenetic origins of cell-to-cell variability were observed in“fractional killing” of tumor cells after exposure to chemotherapy (16).In short,it is almost impossible for cytotoxic therapies to get rid of all the tumor cells as expected.
Dying cells derived from cytotoxic therapy induce tumor repopulation
Under cytotoxic therapy with MTD,it is inevitable to generate massive dying tumor cells.However,the chemoradiation-caused dying tumor cells are not dying silently,these cells undergo significant signaling changes and secret large amounts of molecules to greatly modulate TME and further induce accelerated tumor repopulation,which is the main course of treatment failure (17,18).For example,dying tumor cell-released exosomes promoted the survival and recovery of tumor stem cells (19).Cleaved caspase-3 in apoptotic cancer cells activated calciumindependent phospholipase A2 (iPLA2)/cyclooxygenase 2(COX2)/prostaglandin E2 (PGE2) signaling and further accelerated the proliferation of residual living cells (20).Elevated secretion of PGE2 and vascular endothelial growth factor A (VEGF-A) from dying tumor cells significantly promoted angiogenesis (21),which may support the fast proliferation and repopulation of surviving cancer cells.Moreover,dysregulated microRNA (miRNA)in irradiated dying tumor cells accelerated cancer cell metastasis (22).Altogether,chemoradiation-caused dying tumor cells is a prime motivator of tumor repopulation,which leads to tumor recurrence and therapy failure (17).
Host-mediated responses foster pro-tumorigenic microenvironment
Besides targeting cancer cells,cytotoxic therapies also significantly influence the host normal cells,and the therapy-induced host-mediated local and systemic responses promote establishment of a more tumorsupporting microenvironment (23).The pro-tumorigenic host responses include the accumulation of immune and other stroma cells,and the pro-tumor secretions thereof,which further induce immunological,angiogenic and metastatic effects,etc.It was reported that chemotherapy induced Wnt family member 16B (WNT16B) expression in a therapy-induced DNA damage dependent manner in TME.The elevated WNT16B expression in TME activated the canonical Wnt signaling in tumor cells and conferred them chemoresistance property and promoted disease progression (24).Moreover,gemcitabine and 5-fluorouracil activated inflammasome in myeloid-derived suppressor cells (MDSCs),which then secreted interleukin-1β (IL-1β) to suppress anti-cancer immunity and induced secretion of interleukin-17 (IL-17) in cluster of differentiation antigen 4 (CD4) T cells to promote chemoresistance (25).Upon treatment of tumor-bearing mice with vascular disrupting agents,the circulating endothelial progenitor cells would be homing to the viable tumor rim and potentiated angiogenesis and tumor progression (26).Furthermore,chemotherapy agents including cisplatin and paclitaxel promoted the expression of VEGF receptor 1 (VEGFR-1) in endothelial cells,which created a hostable microenvironment for tumor cell homing and enhanced tumor metastasis (27).
Side effects of cytotoxic therapy evoke therapy-related neoplasms
It is known that cytotoxic therapy tends to induce sideeffects,including numerous short-term and long-term side effects.Of note,the therapy-related neoplasm is quite problematic.Incidence of therapy-related myeloid neoplasm has significantly increased in the last decade in America,with 0.13 cases/100,000 population (28).Considering the number of other cancer types and the incidence of secondary malignancies,the therapy-related neoplasm is a considerable burden (29).Cytotoxic therapycaused dying tumor cells could further promoted secondary carcinogenesis (17).Moreover,in addition to inducing gene mutations,cytotoxic therapy has a significant impact on the microenvironment.It was noted that the tissue microenvironment was the determinative force of clonal evolution and carcinogenesis (30).Even in normal tissues without malignant phenotype,high frequency of tumorassociated somatic mutations was presence (31-33).Cancer therapy with radiation,platinum and topoisomerase II inhibitors was observed to conducively select mutations in DNA damage response genes and drove clonal hematopoiesis,which significantly increased the risk of therapy-related myeloid neoplasms (34).In contrast to the unirradiated mammary gland stroma,the irradiated stroma strongly promoted the initiation of breast cancer and fostered the fast growth of tumors (35).
To treat cancer in another way: Beyond killing tumorous cells
These dilemmas of current cancer cytotoxic therapy urge us to realize that cancer is a systematic complex disease,and more aggressive treatments might not lead to better outcomes,but more side effects.With the growing understanding of tumor biology,especially with respect to TME,we have come to realize that the malignant behavior of cancer cells is not inherently unchanged,cancer cells can be educated to become less aggressive (4,8),and TME can also be normalized to achieve promising effects (36,37).These rapid advances in cancer research inspired us that it would be possible to treat cancer in another way,i.e.transforming cancer cells other than rustically killing tumorous cells.
Directly transforming cancer cells
Contrary to the intensive investigations on how normal cells becoming malignant,less attentions are paid on how malignant cells turning into “normal” ones,which is called tumor reversion (38).Actually,if we could induce the reversion of cancer cells and guide them back to normal state,we might control tumors better (39).The revertant cells,which are quite scarce under spontaneous conditions(about 10-6),could be selected by the infection of H1 parvovirus.By comparing the expression profile of revertant cells and normal cells,Tuynderet al.found that the translationally controlled tumor protein (TCTP),a histamine-releasing factor,was significantly downregulated in the revertant cells.Silencing of TCTP greatly enhanced the proportion of revertant cells,making it up to 30% (40,41).Some antihistaminic compounds and pharmacological compounds with a related structure,including sertraline,were found to suppress TCTP and significantly delayed tumor progression (40).A phase I clinical trial has been launched to test the safety and efficacy of combination chemotherapy with sertraline(ClinicalTrials.gov identifier: NCT02891278).Moreover,the tumor reversion also largely regulated by the microenvironment (42),indicating that reprogramming TME could control tumors better,which will be discussed in the following section.
Differentiation therapy is a successful attempt in transforming tumor cells (4).Application of all-trans retinoic acid (ATRA) to induce differentiation of acute promyelocytic leukemia (APL) cells dramatically improved the clinical cure rate of APL (43).Even more,ATRA was also found to induce glioma tumor cell differentiation to achieve anti-migratory,anti-angiogenic,and therapysensitizing effectsin vitroand impaired the tumor-initiating capacity of these cellsin vivo(44).The histone deacetylase(HDAC) inhibitor,valproic acid,was found to induce rapid terminal granulocytic differentiation of APL cells,and contributed to long-term tumor control (45).Besides,it was reported that methyl sulfone could induce the differentiation of metastatic melanoma cell and suppress the malignant behavior of cancer cells even if the underlying mechanisms remained unclear (46).Vismodegib,a Smoothened inhibitor,was also found to mediate basal cell carcinoma regression by inhibiting a hair follicle-like fate and promoting the differentiation of tumor cells (47).Recently,the histone H3 lysine 9-specific methyltransferase SET domain bifurcated histone lysine methyltransferase 1 (SETDB1) was identified as a key regulator in colorectal cancer by network inference analysis,and inhibition of SETDB1 was found to convert stem-like colorectal cancer cells into postmitotic cells and restore normal morphology (48).
Trans-differentiation expands the field of tumor differentiation therapy.It was reported that combination of MEK (MAP2K7,mitogen-activated protein kinase kinase 7) inhibitors with the anti-diabetic drug rosiglitazone in breast cancer would provoke transdifferentiation of epithelial-mesenchymal transition (EMT)-derived cancer cells into post-mitotic adipocytes,and thus repressing primary tumor invasion and metastasis formation (49).
With advances in biomedical sciences and in-depth understanding of tumor cell biology,efforts were made to reprogram malignant cancer cells toward a benign phenotype.It was reported that inhibition of Nodal (an embryonic morphogen) signaling could promote the reversion of aggressive melanoma cells toward a melanocytic phenotype,and reduce melanoma cell invasiveness and tumorigenicity (50,51).Zhouet al.found that human embryonic stem cell-derived exosomes could reprogram malignant cancer cells to a benign stage and reduce the tumorigenicity (52).Coordinated application of transcription factors hepatocyte nuclear factor 1-α(HNF1A),HNF4A and forkhead box A3 (FOXA3) was found to synergistically reprogram hepatocellular carcinoma cells into hepatocyte-like cells,which lost the malignant phenotypes and retrieved the hepatocyte-specific characteristics (7).Of note,overexpression of GREM1(gremlin 1,DAN family BMP antagonist),a bone morphogenetic protein (BMP) inhibitor,could dramatically suppress pancreatic cancer by inducing complete“epithelialization” of highly mesenchymal pancreatic cancer cells (53).Actually,some lineage-specific transcription factors,combinatorial small molecules,and microRNAs were also applied to reprogram cancer cell from malignancy to benignity (54).
In recent years,inducing tumor cell dormancy has been thought to be an opportunity to outmaneuver cancer (55).Loss of EGFR signaling was revealed to cause senescencelike dormancy in EGFR-mutant lung cancer (56).An agonist of nuclear receptor subfamily 2 group F member 1(NR2F1) was reported to specifically activate dormancy programs in malignant cells,and inhibit lung head and neck squamous cell carcinoma (HNSCC) metastasis (57).
No matter whether guiding cancer cell differentiation,trans-differentiation or reprogramming,or inducing cancer cell reversion or dormancy (Table 1),it does open a window for directly transforming cancer cells.
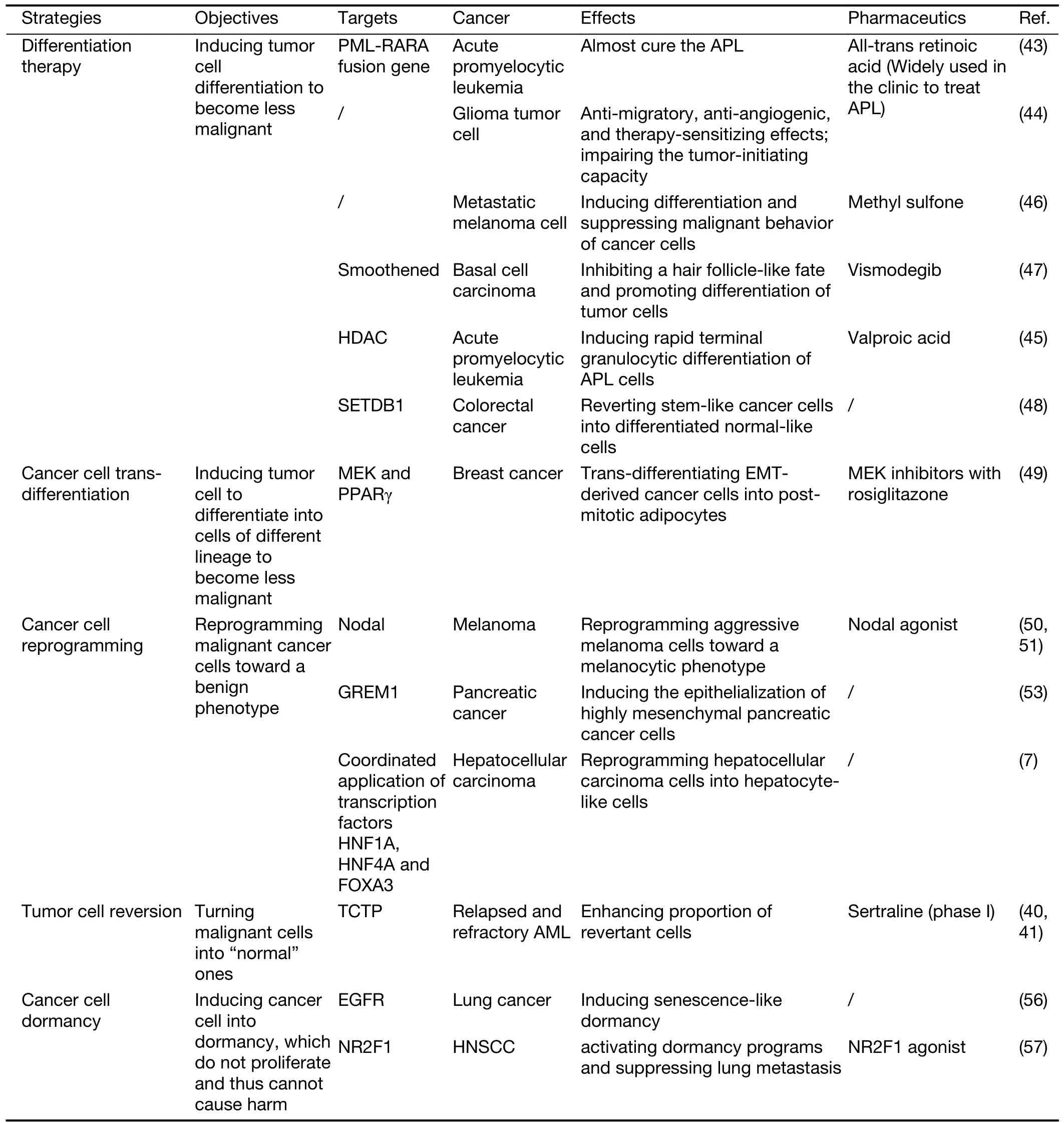
Table 1 Some emerging strategies of directly transforming cancer cells
Transforming TME
Nowadays it is well-known that TME profoundly influences the biological behavior and fate of cancer cells.Generally,the microenvironment around tumor cells is considered pro-tumorigenic (58).But actually,normal tissue microenvironment is generally tumor suppressive,and the interactions between cancer cells and the components of TME are not always mutually promoting,they also mutually restrict to a certain extent (59).As a matter of fact,the tissue microenvironment is largely involved in determining the fate of cancer cells.
Accumulating studies have shown that somatic mutations are not the exclusive oncogenic drivers,the tissue microenvironment,as a determinative force of clonal evolution,is closely related to carcinogenesis (30).Cell-cell contact interactions were supposed to conditionally determine suppression,and incipient neoplastic cells would lie dormant and could persist in the organism indefinitely without carcinogenesis in normal tissues (60).Neoplastic transformation of the mammary epithelial cells could occur only when the stroma was exposed to the chemical carcinogen N-nitrosomethylurea (NMU),regardless of whether or not the epithelial cells were exposed to NMU(61).Irradiated mammary gland stroma was also observed to promote the tumorigenic potential of unirradiated epithelial cells (35).Genetically modified human stroma cells was proved to induce the outgrowth of benign and malignant lesions in ostensibly normal human mammary epithelial cells (62).Genetic deletion of tumor suppressor adenomatous polyposis coli (APC) in murine uterine stroma cells was also shown to induce endometrial hyperplasia and endometrial carcinogenesis (63).
Even more,the normal tissue microenvironment might have the potential to guide the normalization of cancer cells.It was reported that the malignant mouse teratocarcinoma cells were injected into blastocysts,causing them reversal to normalcy and producing normal genetically mosaic mice (64).The normal mammary microenvironment,comprised of stromal,epithelial and host-mediated signals,was also revealed to suppress the tumorigenic phenotype of mouse mammary tumor virus(MMTV)-neu-transformed mammary tumor cells (65),and redirected cancer cells of different species toward normal mammary epithelial cell fate during gland regeneration(65,66).Pancreatic acinar carcinoma cells were also proved to display significant acinar cell differentiated state when cultured on rat testicular seminiferous tubular basement membranes (67).Human metastatic melanoma cells could be also reverted the metastatic melanoma phenotype and reprogrammed into a neural crest cell-like phenotype in an embryonic microenvironment (68).
These studies assumed that the microenvironment around tumor cells might play dominant role indetermining the fate and behavior of malignant cells,even if mutated genes present in these cells.So,normalization of the hostile abnormal TME would contribute to treating cancer or improving therapeutic efficacy (69,70),and reprogramming some components in TME could help to counterbalance or even suppress cancer cells (Figure 2).
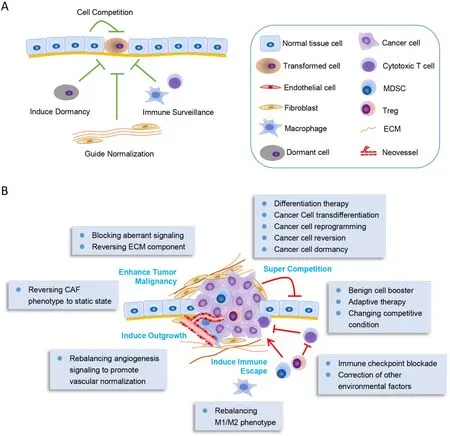
Figure 2 Remodeling tumor microenvironment to transform cancer cells.(A) Normal tissue microenvironment is generally tumor suppressive.The normal cells around the transformed cells would compete with them to suppress their proliferation.The static endothelial cells would induce cell dormancy,and the normal stroma cells as well as ECM would guide normalization of the transformed cells.Moreover,the immune system would exert their immune surveillance function to clear the malignant cells;(B) In advanced cancers,tumor microenvironment is induced to inversely support cancer progression.Inducing cancer cell differentiation or normalizing tumor microenvironment could transform cancer cells to be less malignant.The tumor microenvironment remodeling methods are depicted in the box.In general,these methods mainly consist of inducing normalization of tumor microenvironment or blocking the key transformed signalings.MDSC,myeloid-derived suppressor cell;ECM,extracellular matrix;CAF,cancer-associated fibroblast.
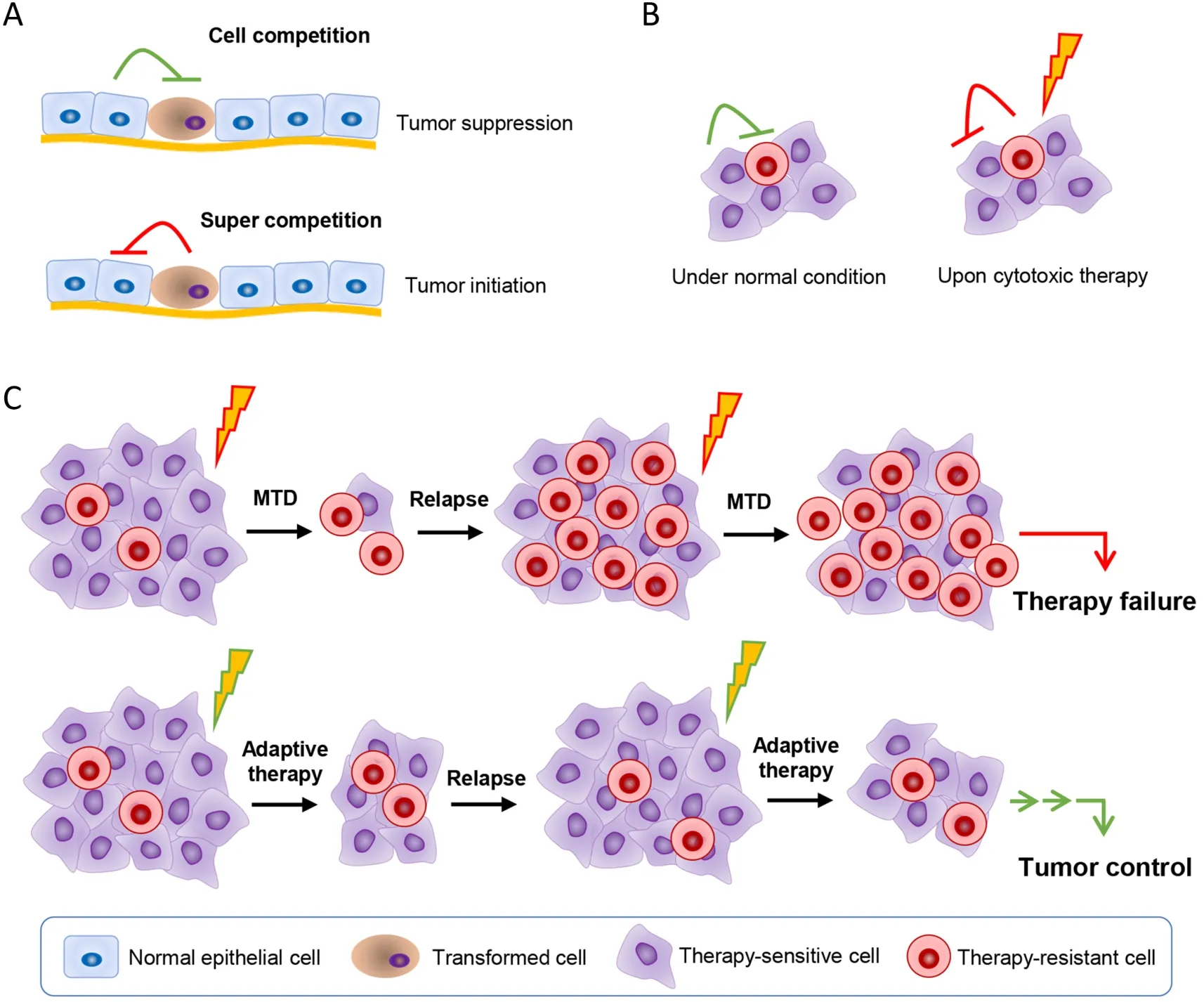
Figure 3 Taking advantage of cell competition to control cancer.(A) Transformed malignant cells are in competition with the normal tissue cells.The transformed cells are generally in the disadvantage position.However,when the normal tissue cells are damaged,or the transformed cells get some mutation that enhance their competition ability,the super competition cells will lead cancer initiation;(B)Cancer cells show great heterogeneous.Cancer cells also compete with each other for the space,nutrition,etc.,and a subpopulation of them are resistant to the applied therapy.The therapy-sensitive cells are at the advantage position compared with the therapy-resistant cells when there’s no therapy insults,and thus the therapy-sensitive cells may in a way inhibit the therapy-resistant cell;(C) When administrating cytotoxic therapy at the traditional MTD,the sensitive cells are wiped out,leaving the therapy-resistant cells to grow.When tumors relapse,majority of cells are the therapy-resistant cells,and thus showing no response to therapy again.Adaptive therapy takes advantage of cell competition by using limited dose of cytotoxic therapy agents to kill a portion of the therapy-sensitive cells so as to reduce tumor burden,while to leave enough therapy-sensitive cells so as to inhibit the therapy-resistant cells.Thus,when tumors relapse,majority of tumor cells are the therapy-sensitive cells,they will response to therapy again,and showing long-term tumor control with limited sideeffects.MTD,maximum tolerable dose.
Leveraging cell competition
As we know,cells are competing with each other due to limited space and nutrients.Cell competition is immanent between mutated cells and wild cells,or cancer cells and normal host cells,and also among cancer cells,which plays vital roles in tissue development and cancer initiation and progression (71).The competition between cancer cells and neighboring normal cells might also provide a window for neighboring normal cells to win a comparative advantage over cancer cells (72) (Figure 3).Epithelial defense against cancer (EDAC) is just such a striking tumor suppressive process,in which normal cells sense and actively eliminate the neighboring transformed cells via cytoskeletal proteins such as Filamin and Vimentin,and orchestrate the cellular homeostasis in epithelial tissues (73,74).The majority of nascent,microscopic,pre-malignant esophageal tumors were observed to be eliminated through competition with mutant clones in the adjacent normal epithelium,with no indication of tumor cell death,decreased proliferation or an anti-tumor immune response in a mouse model (75).Through enhancing the viability of the surrounding normal host cells,the outgrowth of the mutant tumor cells could be constrained (76).In silicosimulations revealed that benign cell boosters,which targeted characteristics of the benign cells and gave a competitive advantage to them,were effective at destroying malignant cells and preventing relapse (77).Specifically enhancing the Hippo signaling in peritumor normal cells would make them outcompete cancer cells,contributing to liver cancer regression in mice(59).Antioxidant pretreatment was revealed to improve normal cell fitness and outcompete p53-mutant cells caused by the low-dose ionizing radiation (LDIR) in the transgenic mouse esophagus (78).It was reported that simply changing the intraprostatic pH by adding NaHCO3to the drinking water could alter the competitive advantage position and greatly diminished growth of primary and metastatic tumors (79).
However,the mutant malignant cells,such as APCmutant intestinal stem cells (ISCs),could work as supercompetitors to actively suppress the neighboring wild-type counterparts by secreting WNT antagonists such as NOTUM (palmitoleoyl-protein carboxylesterase NOTUM),and established the competitive advantage.Yet,blocking the inhibitory effect of APC-mutant cells with lithium chloride,which rendered wild-type ISCs insensitive to WNT antagonists,or directly inhibition of NOTUM,limited the expansion of pre-malignant clones (80,81).These attempts open a window for further efforts.
Actually,the intra-tumoral competition among cancer cells is also immanent (82).It was noted that the therapyresistant cells would pay a price to reduce cell fitness for evolving resistance to chemotherapy or targeted therapies(8).So,adaptive therapy was accordingly proposed,which enforces a stable tumor mass rather than eradicates all cancer cells (83).Adaptive therapy typically gives progressively lower doses of cytotoxic drugs rather than MTD used in conventional chemotherapy.In that,only a small portion of tumor cells are killed to control tumor mass,yet a significant population of chemo-sensitive cells are preserved to survive so as to suppress proliferation of the less fit but chemo-resistant subpopulations in turn (8).That is to say that adaptive therapy enables the therapysensitive cells to outcompete the therapy-resistant cells.In fact,this modality has significantly enhanced progressionfree survival in many types of cancers.In breast cancer mouse models,compared with standard high-dose-density treatment,low doses of verapamil and 2-deoxyglucose,which accentuate the cost of resistance and decrease energy production,were found to suppress the proliferation of drug-resistant clonesin vivo,leading to 2-to 10-fold increase in progression-free survival (84).Besides,treatment cycles determined by patient-specific tumor dynamics rather than fixed cycles were revealed to suppress proliferation of androgen-independent cells and lowered cumulative drug dose in the metastatic castrate-resistant prostate cancer (85).In a model of resistance to cyclindependent kinase inhibitor (CDKi),low-dose CDKi outperformed high-dose CDKi in controlling tumor burden and therapy resistance in tumor spheroids due to that the resistant cells reduced the proliferative fitness (86).
Reprogramming the TME
Cancer-associated fibroblasts (CAFs),one of the main components of TME,are educated or reprogrammed by cancer cells,and are usually thought to potentiate cancer progression (87).However,the normal tissue fibroblasts,the major source of CAFs,are inherently tumor suppressive(88).But once the fibroblasts are activated or reprogrammed to CAF,they may conversely promote tumor progression.Thus,depletion of CAFs was adopted to suppress cancer progression (89).But inconceivably,two independent studies simultaneously reported that depleting CAFs,either by depleting sonic hedgehog or αSMA+(actin alpha 2,smooth muscle) myofibroblast-depletion,conversely promoted pancreatic cancer progression (90,91).These facts remind us that targeting CAF to block or inhibit tumor progression is not a simple task (92).Actually,fibroblasts are highly heterogeneous,different subtypes of CAFs can exert different functions,and some subtypes of CAFs have the potentials to inhibit cancer cells(93).Recently,it was revealed that specifically depleting collagen I in αSMA+myofibroblasts promoted pancreatic cancer initiation and progression partly via immune suppression,indicating that αSMA+myofibroblast-produced collagen I restrains cancer progression (94).In that,reprogramming CAFs or harnessing a sort of CAFs to control cancer might be a potential way.Some efforts are made to reprogram CAFs to a more static state resembling normal fibroblasts for evoking the tumor suppressive potentials.Calcipotriol,a vitamin D analog,was used to reprogram CAFs to a less inflammatory,quiescent state,leading to less tumor aggressive phenotype and better survival (95).ATRA was also revealed to induce quiescence and reduced motility of pancreatic stellate cells by affecting paracrine Wnt/β-catenin signaling in pancreatic cancer,thereby inhibiting cancer cells and altering tumor morphology in LSL-KrasG12D/LSL-Trp53R172H/Pdx1-Cre(KPC) mice (96).Losartan,an angiotensin antagonist,could down-regulate the expression of profibrotic signals transforming growth factor β 1 (TGF-β1),CCN family member 2 (CCN2) and endothelin-1 (ET-1),and thus reducing stromal collagen and hyaluronan production,leading to better chemotherapy efficacy (97).Angiotensin receptor blockers were also reported to convert myofibroblast CAFs to an immune-supportive state and improve T lymphocyte activity,dramatically enhancing the efficacy of immune checkpoint blockade immunotherapy in desmoplastic breast cancers (98).These evidences strongly suggest that reprograming of CAFs is of help to tumor control.
Tumor-associated macrophages (TAMs) are also locally educated by cancer cells to express distinct transcriptional landscapes and possess an alternatively activated (or M2)phenotype (99),which entails immunosuppression and tumor-promotion.Nevertheless,the M1 phenotype is intrinsically tumor suppressive.Thus,reprogramming the M2 macrophages to M1 phenotype has attracted extensive attentions.Macrophage colony-stimulating factor (CSF-1)is a vital factor for the recruitment,survival and M2-like differentiation of TAMs.Treatment with anti-CSF-1R(CSF-1 receptor) monoclonal antibody (RG7155) strongly reduced CSF1R+CD163+macrophages in tumor tissues,contributing to clinical objective responses in diffuse-type giant cell tumor patients (100).An orally active CSF-1R inhibitor,JNJ-28312141,was also found to reduce F4/80+TAMs or tumor-associated osteoclasts and prolong the survival of tumor-bearing mice (101).CSF-1R inhibition was also revealed to significantly block glioma progression by altering gene signatures of TAMs and decreasing expression of M2 phenotype (102).Moreover,combination of CD40 agonists and anti-CSF1 antibody led to profound TAM reprogramming and created pro-inflammatory environment for effective T cell responses (103).Recently,blockade of CD24 or its inhibitory receptor sialic-acidbinding Ig-like lectin 10 (Siglec-10) was reported to dramatically unleash the phagocytic role of macrophages and promoted tumor regression in ovarian cancer and triple-negative breast cancer (104).Besides,the macrophage receptor macrophage receptor with collagenous structure (MARCO) defined a subtype of immune suppressive TAMs.Treatment with anti-MARCO monoclonal antibody could induce anti-tumor activity through reprogramming TAM populations to a proinflammatory phenotype and increasing tumor immunogenicity (105).Activating signaling in TAMs with STING(stimulator of interferon genes protein) agonists reprogrammed M2-like pro-tumor macrophages into an M1-like anti-tumor state,which enhanced the sensitivity of poly (ADP-ribose) polymerase 1 (PARP) inhibitor in breast cancer susceptibility protein (BRCA) mutant breast cancer by elevating the anti-tumor immunity (106).Furthermore,targeting the aberrant epigenetic signaling also contributes to TAM reprogramming (107).
Tumor vasculature is characterized as disorganized,immature and permeable,which contributes to establishing hypoxic microenvironment and promoting cancer cell metastasis (108).Targeting tumor angiogenesis has long been used for cancer treatment.However,the effects of large dose anti-angiogenesis agents were unsatisfactory and even promoted cancer progression and metastasis (109).Hence,vascular normalization has become an alternative strategy and shown therapeutic potentials (108,110).Appropriate VEGFR-2 blockade created a “normalization window”,which could normalize tumor vessels,relieve tumor hypoxia and improve tumor response to radiation(111).Lower dose of anti-angiogenesis agents rather than higher one would normalize angiogenic microenvironment,which further facilitated immune microenvironment normalization (112).Combined tumor angiogenesis inhibition and tumor vessel stabilization by an Ang2-binding and Tie2-activating antibody was revealed to normalize tumor vessels,promote blood perfusion and further improve immune cell infiltrations,and resulted in better tumor control without notable vascular and hematologic adverse effects in several tumor models (37).Moreover,Chloroquine was found to normalize tumor vessels via influencing endosomal Notch1 trafficking,independent of autophagy (113).Recently,activating sphingosine 1-phosphate receptor was revealed to promote normalization of tumor vessels,which suppress tumor growth and metastasis (114).Additionally,cross-talk between tumor cells and activated endothelial cells in“vascular niche” was reported to trigger cancer cell escape from dormancy via the Notch3-Dll4 interaction in T cell acute lymphoblastic leukemia (T-ALL) cells or colorectal cancer (115).In breast cancer patients,the static endothelium-derived thrombospondin-1 was revealed to induce sustained cell quiescence (116).These facts indicated that inducing dormancy of endothelial cells would contribute to suppressing cancer progression.
Unleashing the potential of anti-tumor immunity have long been the pursuit in cancer therapy.However,there was no big improvement before the discovery of immune checkpoint blockade therapy including programmed cell death 1 ligand 1 (PD-L1) and cytotoxic T-lymphocyte protein 4 (CTLA-4) blockade therapy,which is committed to establish immune normalization by blocking the aberrant immune inhibitory signaling (36).However,the tumor immune microenvironment (TIME) is complex,and targeting a single molecule can benefit a certain population of cancer patients,but most patients still do not respond well to immune checkpoint blockade therapy (117).Recently,a critical immune suppressor Siglec-15,whose expression is mutually exclusive to B7-H1 (PD-L1) and is broadly up-regulated in human cancer cells and tumorinfiltrating myeloid cells,was identified as a potential target for normalization cancer immunotherapy (118).Similarly,CD161 was also identified as an inhibitory receptor,and blocking of which enhanced the T cell-mediated killing of glioma cellsin vitroand anti-tumor functionin vivo(119).Remodeling other immune cells also offers therapeutic opportunities.Noteworthily,loss of T cell immunoglobulin and mucin-containing molecule 3 (TIM-3) in dendritic cells (DCs) but not in CD4+or CD8+T cells unleashed strong anti-tumor immunity by regulating inflammasome activation (120).Adjuvant epigenetic therapy with low-dose DNA methyltransferase and histone deacetylase inhibitors,5-azacytidine and entinostat,disrupted the premetastatic microenvironment and inhibited lung metastases by inhibiting myeloid-derived suppressor cell (MDSC) trafficking and promoting MDSC differentiation (121).The immunosuppressive regulatory T cells (Tregs) could also be reprogrammed into a proinflammatory phenotype under certain acute proinflammatory signals,and thus enhancing anti-tumor immune responses (122).
The dense and aberrant extracellular matrix (ECM)around cancer cells also greatly influences tumorigenesis,cancer progression and metastasis,and contributes to the heterogeneous distribution of chemotherapeutic agents,which makes certain areas of solid tumors difficult to treat(123).As the direct contact microenvironment of tumor cells,ECM plays a vital role in regulating cell biology.It was reported that embryonic mesenchyme from early stage(E12.5-13.5) mammary glands could induce partial breast cancer reversion or normalization (124),implying that ECM normalization would contribute to tumor control.Actually,about two decades ago,Weaveret al.reported that treatment of breast cancer cells with β1-integrin blocking antibody could morphologically and functionally reverse their malignant phenotype in 3D culture and nude mice (125).Later on,integrin β1 neutralizing antibody was found to inhibit the switch from tumor cell dormancy to proliferative metastatic growth,leading to significantly reduced metastatic outgrowth in breast cancer (126).Moreover,laminin-1 was found to uniquely express in normal breast myoepithelial cells,and replenishment of laminin-1 would impart polarity and basement membrane deposition of luminal breast epithelial cells,and reconstituted normal structures of double-layered breast acini with central lumina in breast tumor tissues (127).Moreover,certain ECM component in the TME was revealed to constrain cancer progression.Type I collagen,the main component of ECM,was reported to mechanically restrain tumor spread,overrode its own stiffness-induced mechano-signals (94,128),indicating that the function of ECM is complex and need further investigations.
Remarkably,microenvironment-targeting combination strategies,which take into account the interplay between cancer cells,immune cells,stroma and the vasculature,were developed to improve cancer immunotherapy (117).Combination treatment of immunotherapy with stroma and/or vascular normalization decreased tumor growth rate and increased the efficacy of immunotherapy in various cancers (129).And combined use of programmed cell death protein 1 (PD-1) and low-dose VEGFR-2 blockade was demonstrated to simultaneously normalize immune and angiogenic microenvironment,which significantly promoted tumor shrinkage and enhanced survival of hepatocellular carcinoma mice (130).Blockade of Tim-4,a receptor for phosphatidylserine (PS),in cavity-resident macrophages was also revealed to potentiate anti-tumor immunity and enhanced the efficacy of anti-PD-1 therapy and adoptive T cell therapy in mice (131).
Conclusion remarks and future perspectives
Herein,we demonstrated that cancer cells can barely be wiped out by cytotoxic therapy,which would further induce tumor-promoting responses and many side-effects.After thoroughly interrogating the biology of cancer,we proposed that transforming cancer cells could be an acceptable approach to achieve long-term cancer control.We demonstrated that directly transforming cancer cells shows promising effects.More importantly,TME plays a vital role in determining the fate of cancer cells,and the deteriorated microenvironment actively promotes cancer initiation and progression.Hence,leveraging cell competition to outcompete malignant cells also shows therapeutic potentials,and remodeling TME,including reprogramming CAFs,TAMs and ECM,normalizing angiogenic and immune microenvironment,can guide malignant cells back to more benign phenotype and achieve long-term control without causing significant side-effects,even if the “normalization window” may be not wide,the normalizing agents may be limited.Moreover,simultaneously remodeling more than one component in the TME may show synergistically effects,such as normalizing tumor vessels and TIME as aforementioned.
However,some questions remain to be answered.We should note that most strategies aimed at transforming cancer cells have not been extensively tested in the clinic,they have just been proven in pre-clinical studies.Thus,more clinical investigations are highly needed.Currently,differentiation therapy in acute myeloid leukemia and immune checkpoint blockade therapy in certain cancers have achieved great success in the clinic,and adaptive therapy have been tested to be more effective in several cancers (8,36,79).Another challenge is that cancer biology is so complex that we still know little about it.Despite tremendous advances in tumor biology,there are still many mysteries to explore,especially on cancer cellmicroenvironment communications and their biological effects.Thus,more extensive bench works are needed and more effective experimental models are greatly required.Advances in biomedical technologies,such as spatial omics and multiplexed imaging,single-cell profiling and lineage tracing,multiparameter flow cytometry and imaging mass cytometry,and novelin vitroandin vivocancer models,e.g.3D cell culture,patient-derived organoids (PDO) and patient-derived xenografts (PDX),etc.,will greatly facilitate the investigation of cancer biology (132).Furthermore,high-quality meta-analyses would provide more comprehensive insights on complex biology (133).
Certainly,we hereinabove do not intend to repudiate cytotoxic therapy.On the contrary,we would like to replenish their deficiency or even bridge them.It was noted that radiation therapy could remodel TIME by activating immune-stimulatory as well as immune-suppressive signalings and potentiated an immune-permissive TME(134).It was believed that triggering tumor-localized inflammatory forms of cell death,such as necroptosis and pyroptosis,might contribute to converting these immune“cold” tumors to “hot” tumors and alter the TIME toward an immune checkpoint inhibitor (ICI)-responsive state(135,136).Therapy-induced modulation of the TME might potentially represent a novel way to enhance local and systemic treatments (137).
In conclusion,for better tumor treatment and long-term cancer control,new therapeutic strategies beyond cytotoxic therapy should be accordingly developed.With the deep understanding of tumor biology,especially on TME,it would be a feasible way to transform cancer cells for longterm living with cancer.
Acknowledgements
This study was supported by the funding of the National Natural Science Foundation of China (No.82073203) and Guangdong Basic and Applied Basic Research Foundation(No.2023A1515012903 and 2022A1515110033).
Footnote
Conflicts of Interest: The authors have no conflicts of interest to declare.
 Chinese Journal of Cancer Research2023年2期
Chinese Journal of Cancer Research2023年2期
- Chinese Journal of Cancer Research的其它文章
- Stomach cancer burden in China: Epidemiology and prevention
- When immunotherapy meets liver transplantation for hepatocellular carcinoma: A bumpy but promising road
- Integrated strategies for chemotherapy cycles in nasopharyngeal carcinoma patients: Real-world data from two epidemic centers guiding decision-making
- Variant rs8400 enhances ALKBH5 expression through disrupting miR-186 binding and promotes neuroblastoma progression
- Exploration and optimization of surgical techniques for laparoscopic transhiatal lower mediastinal lymph node dissection for adenocarcinoma of esophagogastric junction: A prospective IDEAL 2a study with qualitative design
- Aqueous-soluble components of sporoderm-removed Ganoderma lucidum spore powder promote ferroptosis in oral squamous cell carcinoma