
慢性镉暴露下食管鳞状细胞癌细胞的lncRNAmRNA共表达网络分析
2023-08-07 04:20曾明钦陈炯玉张厦蓉黄裔腾
癌变·畸变·突变 2023年4期
彭 琳,曾明钦,陈炯玉,张厦蓉,朱 芮,黄裔腾
(1.汕头大学医学院附属肿瘤医院中心实验室,广东 汕头 515041;2.汕头大学医学院附属第一医院健康管理中心,广东 汕头 515041)
食管癌是常见的消化道恶性肿瘤,位列全球恶性肿瘤发病率第7位和死亡率第6位[1]。鳞状细胞癌和腺癌是食管癌的两种主要组织学类型,其中食管鳞状细胞癌(esophageal squamous cell carcinoma,ESCC)主要高发于东亚男性,尤其在中国约占食管癌病例的90%[2-3]。ESCC 具有的明显地域、性别差异的流行病学特征[4],高度提示其病因学的特殊性,环境因素被认为是ESCC发生的重要危险因素[5]。镉是环境中广泛存在的重金属污染物,职业人群镉暴露主要通过呼吸道吸入和皮肤暴露,一般人群主要通过空气、吸烟、食物和水等途径,其中,膳食摄入是日常暴露的主要来源,如稻谷、水产品、马铃薯、蔬菜、肉类和海产品等[6-7]。调查显示,中国男性和女性成年人平均每周饮食镉摄入量分别为3.9和4.1 μg/kg[8],约为欧洲国家人均每周饮食镉暴露水平(2.04 μg/kg)的2 倍[9]。近年来,已有若干基于中国人群的流行病学证据指出镉暴露与食管癌的关联[10-12]。本课题组前期在我国食管癌高发唯一沿海地区——潮汕地区开展的人群研究也表明,血镉负荷是ESCC 发生和不良预后的危险因素之一,且通过构建持续低剂量镉暴露人ESCC 细胞株(CCT-EC109和CCT-EC9706)的系列体内外实验证明慢性低浓度镉暴露促进ESCC 细胞生长、侵袭、迁移能力,增强肿瘤干细胞特性和对放化疗耐受性[13],但具体机制有待深入解析。
镉是非直接遗传毒性致癌物,表观遗传途径是其主要致癌机制[14]。长链非编码RNA(long non-coding RNA,lncRNA)是一类长度超过200 nt 的无蛋白编码功能的RNA。新近研究指出,镉的毒性效应存在组织器官特异性,可能与其调控不同lncRNA 激活相应的信号通路或分子事件有关[15]。然而,lncRNA 在镉促ESCC 中的调控机制尚无报道。本文拟基于转录组测序技术,利用课题组前期建立的慢性镉暴露ESCC 细胞模型之一CCT-EC109细胞系,通过比较其与亲本细胞EC109 在lncRNA 和mRNA 水平的表达差异,运用生物信息学方法构建lncRNA-mRNA 共表达网络,为探讨慢性镉暴露促进ESCC 演进相关机制研究提供基础。
1 材料与方法
1.1 细胞模型构建
EC109 细胞由汕头大学医学院附属肿瘤医院中心实验室细胞库提供,在37 ℃、CO2体积分数为5%条件下培养于含10%胎牛血清和含青-链霉素的RPIM-1640 培养基中。EC109 细胞经含5 μmol/L 氯化镉的培养基持续培养12周,建立慢性镉暴露细胞模型CCT-EC109。
1.2 主要试剂
氯化镉(纯度99%)购自美国Sigma 公司,RPMI-1640 培养基和10%胎牛血清均购自美国Gibco,青霉素/链霉素、0.25% EDTA 胰蛋白酶均购于北京索莱宝科技有限公司,RNA later 购自美国Invitrogen。细胞总RNA提取试剂盒购自德国Qiagen公司;逆转录试剂盒购自南京诺唯赞医疗科技有限公司;实时荧光定量PCR(quantitative real time PCR,qPCR)试剂盒购自上海翌圣生物科技股份有限公司。
1.3 总RNA提取及细胞转录组测序
EC109 和CCT-EC109 两株细胞各收集3 个样本,使用miRNeasy Mini 试剂盒(德国Qiagen)抽提总RNA,使用Qubit®3.0 Fluorometer 和NanoDrop One 分光光度计定量,并利用Agilent Bioanalyzer 2100 选择RNA 完整性指数值大于7.0的样品用于测序建库,1 μg RNA用于后续RNA样品制备。建库测序委托上海鲸舟基因科技有限公司完成,lncRNA 和mRNA 测序采用Illumina NovaSeq 6000 测序仪,质控标准: 每个样本数据量约10 G,所有类型碱基质量大于20 的比例不小于90%。
1.4 差异表达lncRNA和mRNA的筛选
使用Hisat 2.0.5 将测序后的原始数据匹配到参考基因组,将输出的序列比对/图文件转换为二进制比对/图文件,并使用SAMtools 1.3.1 进行排序。使用R软件包进行分位数归一化以及后续数据处理,以每千个碱基的转录每百万映射读取的片段数表示基因丰度,利用Stringtie 软件对每个基因中的片段进行计数,并根据TMM算法进行归一化。使用R包edgeR进行mRNA和lncRNA的差异表达分析,筛选条件为:P<0.05,|log2(fold change)|≥1。
1.5 差异lncRNA相关靶基因筛选
应用ENCORI(http://starbase.sysu.edu.cn) 对DElncRNA 进行顺式与反式靶基因预测,继而利用RNAplex 软件计算lncRNA-mRNA 互补配对热动力学参数值,根据软件阈值进一步筛选靶基因。最后,将DE-lncRNA 预测的靶基因集与DE-mRNA 基因集取交集,获得DE-lncRNA调控的镉暴露相关基因。
1.6 功能预测
为进一步了解DE-lncRNA调控的靶基因功能,利用R包Enrich进行基因本体(gene ontology,GO)功能注释,同时采用京都基因与基因组大百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)数据库(http://www.genome.ad.jp/kegg),进行通路富集分析。
1.7 LncRNA-mRNA共表达网络构建
根据1.5 筛选的lncRNA 相关靶基因结果,采用Pearson's 相关分析筛选相关系数≥0.9(P<0.05)的结果,利用Cytoscape 软件构建lncRNA-mRNA关系对。
1.8 qPCR 验证共表达网络关键lncRNA 和mRNA 的表达
提取EC109、CCT-EC109 细胞总RNA,利用HiScript®III cDNA 第1 链合成试剂盒和HiScript®III RT SuperMix for qPCR 试剂盒(诺唯赞,中国)分别将lncRNA和mRNA逆转录成模板cDNA后,以β-actin为内参,qPCR 测定目的lncRNA 和mRNA。PCR 引物序列见表1,引物由广州艾基生物技术有限公司合成。反应体系: cDNA 模板1 μL,上下游引物各0.4 μL,2×ChamQ Universal SYBR qPCR Master 混合物10 μL,双 蒸 水8.2 μL。使 用Bio-Rad CFX96 Deep Well PCR 仪进行扩增,条件: 预变性95 ℃、30 s;循环反应95 ℃、10 s,60 ℃、30 s,共40 个循环。记录各孔CT值,目的基因的相对表达水平为2-ΔΔCT。每组实验重复3次。

表1 目的lncRNA和mRNA的引物序列
1.9 统计分析
采用R4.0.5 软件和SPSS 24.0 进行统计学分析。组间差异比较采用Student'st检验,P<0.05 表示差异有统计学意义。
2 结 果
2.1 暴露株与亲本的差异lncRNA和mRNA表达谱
层次聚类分析表明,lncRNA和mRNA表达谱可较明显区分暴露株和亲本株(图1)。散点图可视化显示,2 794个DE-lncRNA包含1 248个上调lncRNA和1 546个下调lncRNA;4 267 个DE-mRNA 则包括2 766 个上调基因和2 947个下调基因(图2)。

图1 EC-109与CCT-EC109细胞lncRNA和mRNA差异表达谱

图2 差异lncRNA相关的靶mRNA
2.2 差异表达lncRNA相关的靶mRNA
如图2 所示,将DE-lncRNA 预测的7 216 个靶基因与4 267个DE-mRNA取交集,筛选得到1 546个与lncRNA相关的靶基因,其中上调基因737个,下调基因810个。
2.3 镉暴露相关lncRNA的靶基因生物学功能分析
将上述筛选的1 546 个交集基因进行GO 聚类分析,结果显示靶基因富含57 个功能项,包括28 个生物学过程项、12个分子功能项和17个细胞成分项。涉及的生物学过程主要包括代谢、生物调节、刺激应答、免疫系统、细胞增殖等;细胞组分方面主要为膜、膜性腔、含蛋白质的复合物等;分子功能项主要与催化活性、转录调节活性、分子功能调节等密切相关(图3)。KEGG 分析1 546 个靶基因富集信号通路包含324 种途径,癌症和信号转导是最主要的条目,其中,最显著的富集项为Hippo 信号通路(富集基因数23,P=0.003)和病毒致癌作用(富集基因数29,P=0.002),其他重要信号通路包括癌症信号通路(富集基因数54,P=0.044)、溶酶体(富集基因数137,P=0.051)等。KEGG显示镉暴露相关lncRNA的靶基因通路富集分析的前30个信号通路见图4。
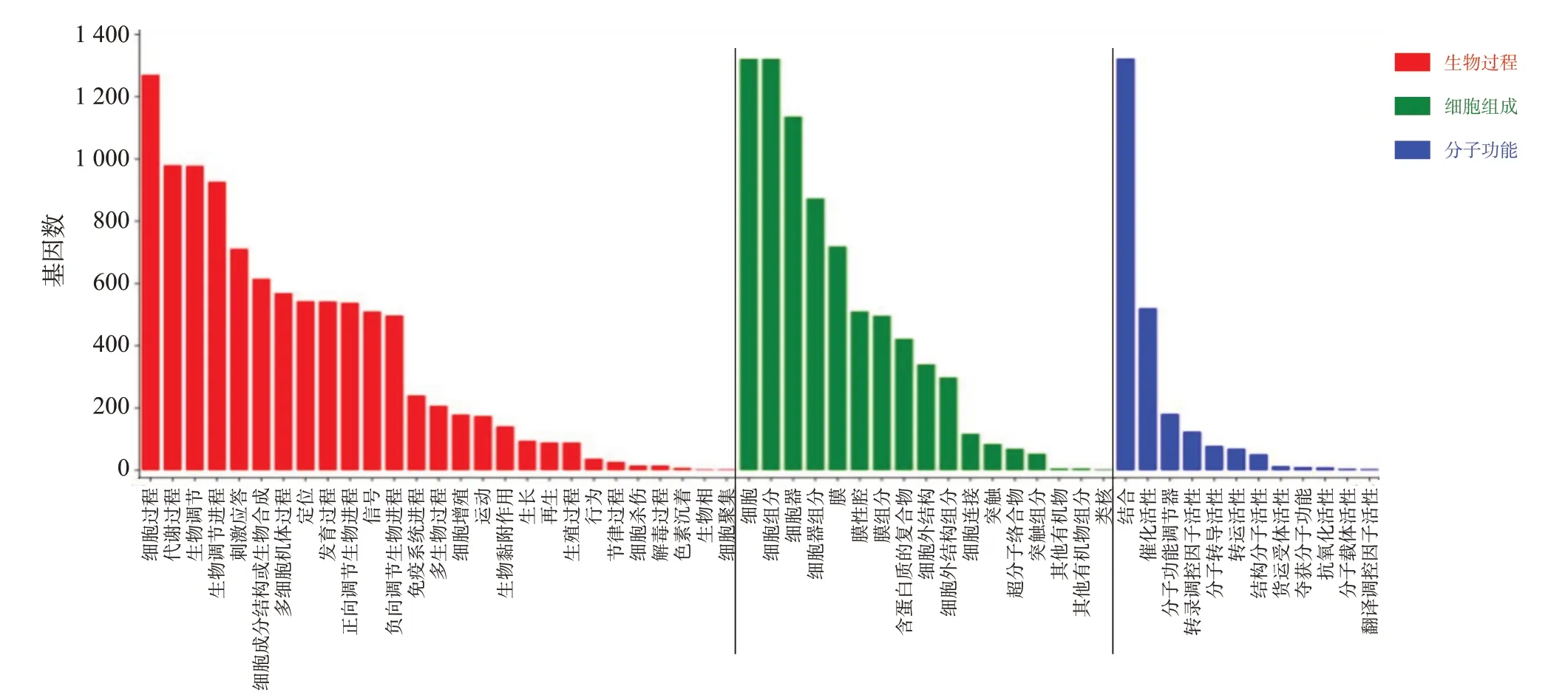
图3 镉暴露相关lncRNA的靶基因GO功能富集分析
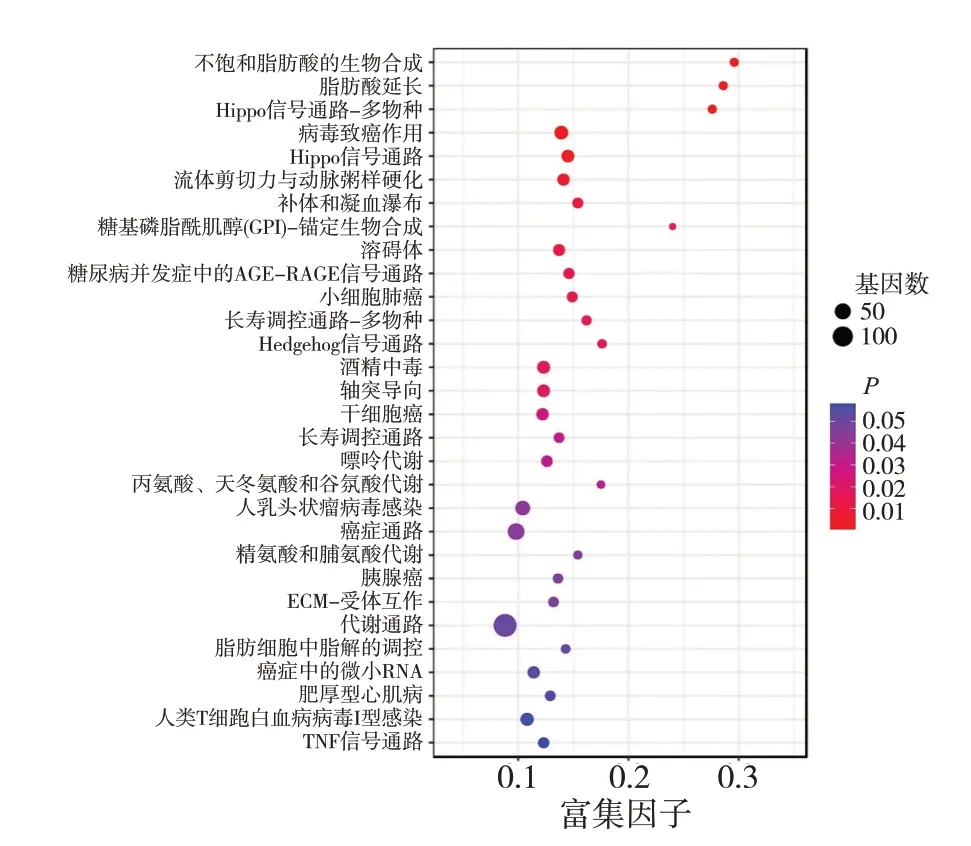
图4 镉暴露相关lncRNA的靶基因通路富集分析
2.4 镉暴露相关lncRNA-mRNA共表达网络
根据Pearson's 相关系数筛选出的前1 000 个lncRNA-mRNA 共表达基因对包含231 个DE-lncRNA和347 个DE-mRNA,Cytoscape 软件构建的共表达网络如图5 所示。将差异表达最显著的前10 个lncRNA(NONHSAT244320.1、NONHSAT071835.2、NONHSAT 225909.1、NONHSAT107780.2、NONHSAT256799.1、NONHSAT097388.2、NONHSAT188800.1、 NONHSAT 211602.1、 NONHSAT217261.1、 NONHSAT054462.2)的38个共表达mRNA进一步构建关系对网络(图6),与mRNA 关联最多的lncRNA 为NONHSAT097388.2,其在暴露株表达显著下调。共13个DE-mRNA 被预测为该lncRNA 的靶基因,分别是EMP2、ORAI2、ECE1、ZNF713、 HGSNAT、 CA5B、 ARPIN、 ANKRD9、PGF、MFAP5、PGM2L1、GNE和FUT8,其中FUT8、MFAP5、PGF显示为上调基因,与NONHSAT097388.2均呈负相关关系。

图5 镉暴露相关前1 000个lncRNA-mRNA共表达关系对

图6 前10个差异表达lncRNA的lncRNA-mRNA共表达网络
2.5 共表达网络相关基因的表达验证
为进一步验证构建的共表达网络中关键lncRNA和mRNA 的表达情况,选取mRNA 关联最多的lncRNA NONHSAT097388.2 及其负相关基因FUT8、PGF、MFAP5和 正 相 关 前4 位 基 因EMP2、ECE1、ORAI1、ZNF713,qPCR 测定结果显示,除FUT8外,其他基因在亲本株EC109与暴露株CCT-EC109的表达差异与转录组测序结果一致,即MFAP5和PGF在暴露株中表达升 高(P<0.05), 而NONHSAT097388.2、ECE1、EMP2、ORAT1和ZNF713表 达 均 下 降(P<0.01),如图7。

图7 qPCR验证镉暴露相关lncRNA-mRNA共表达网络关键基因在EC109和CCT-EC109细胞中的表达
3 讨 论
镉致癌机制复杂,可引起包括DNA链断裂、染色体畸变和基因突变在内的遗传毒性作用,但表观遗传改变被认为是比直接DNA损伤更为主要的致癌分子事件[16]。近年来,lncRNA作为新兴表观遗传调控分子在肿瘤发生发展中的作用成为研究热点。lncRNA不仅参与mRNA 稳定性、衰变调控,还参与基因翻译及翻译后修饰调节等诸多生物学过程[17]。新近研究证据揭示了lncRNA 参与镉的毒性作用,包括多器官损伤、生殖毒性、DNA 损伤修复异常、恶性转化等过程[15,18]。例如,持续镉暴露可诱导前列腺癌细胞lncRNA OIP5-AS1 表达升高,通过miR-128-3p/SLC7A11 轴抑制铁死亡参与前列腺癌进展[19]。lncRNA DUXAP10 通过调控Hedgehog通路参与镉暴露诱导人支气管上皮细胞干细胞特性增加[20]。此外,lncRNA MALAT1也被认为是镉毒性的潜在标志物,它介导镉暴露诱导的人支气管上皮细胞16HBE细胞增殖、侵袭和迁移能力的增强,但具体机制不明[21]。迄今为止,尚无镉暴露对食管癌细胞lncRNA表达谱影响的研究报道。
通常认为,共表达基因集参与同一通路,或者受到同样的调控,具有相似的生物学功能。因此,lncRNA-mRNA 互相作用关联分析方法常被应用于lncRNA在疾病尤其是在肿瘤中的功能和调控机制的研究[22]。本研究首先通过功能及通路富集分析发现,镉暴露ESCC 细胞株发生变化的基因在生物学功能上主要涉及细胞代谢、刺激应答、免疫系统、细胞增殖、转录调控、信号转导等方面,这些也与既往报道阐述的镉毒性作用及相关机制相符[23-24]。尽管关于镉免疫毒性对肿瘤演进影响的直接研究不多,但大量研究表明,镉暴露可影响血液或组织中免疫细胞稳态如Th1/Th2 的失衡和许多免疫相关分子,如IL-2、IL-4、IL-6、IL-10、IFN-γ、TNF-α、IL-1β等的表达发生变化[25-26],这些均可能影响肿瘤免疫微环境。值得一提的是,在镉影响的信号转导通路中,Hippo 通路最显著,而该通路被证实参与食管癌的恶性进展和化疗抵抗机制[27]。可见,Hippo信号通路可能是慢性镉暴露促进ESCC 细胞恶性生物学行为及放化疗抵抗的重要分子机制。此外,通路富集分析还显示,病毒致癌作用也是显著富集项,这与Liang等[28]在镉暴露乳腺癌细胞株富集到HPV感染相关通路的结果相近,提示镉可能与病毒具有协同致癌作用。
对共表达网络关系对分析发现,与mRNA 关联最多的lncRNA NONHSAT097388.2 在暴露细胞株CCT-EC109中显示表达降低,提示镉暴露可能通过抑制该lncRNA 的表达调控相关基因及相应分子事件。NONCODE 网站查询显示,NONHSAT097388.2 基因位于4号染色体,长度14 407 bp,包含6个外显子,目前对NONHSAT097388.2 的功能知之甚少,搜索与其关联的2 个上调基因MFAP5、PGF相关文献,发现尽管MFAP5在食管癌中的作用未见报道,但在头颈部鳞癌和乳腺癌的研究中,MFAP5被证实分别通过AKT通路和ERK/MMP 通路促进肿瘤细胞的上皮间质转化(epithelial-mesenchymal transition,EMT)、增 殖 和 转移[29-31];阻断MFAP5 的抗肿瘤免疫治疗则可增加卵巢癌和胰腺癌对紫杉醇化疗的敏感性[32]。PGF基因即胎盘生长因子(placenta growth factor,PGF/PIGF)基因,是血管内皮细胞生长因子(vascular endothelial growth factor,VEGF)家族重要成员之一。新近研究表明,PGF 在肝内胆管细胞癌细胞和患者血浆中表达增加,且与疾病进展相关,而阻断PGF可抑制肿瘤细胞的侵袭、转移和化疗抵抗[33];PGF 还被发现在恶性程度最高的三阴性乳腺癌组织中的表达水平高于其他亚型,而且,在乳腺癌、黑色素瘤患者的血液中均能检测到高丰度的循环PGF表达;高度提示PGF是促进肿瘤侵袭、转移的重要因素之一[34-35]。EMP2基因是与NONHSAT097388.2 共表达的正相关基因之一,EMP2 mRNA 被证实在食管癌组织中显著低表达,且在淋巴结阳性和中低分化组中表达水平更低[36]。上述结果提示, 镉暴露可能通过NONHSAT097388.2 介导MFAP5、PGF、EMP2等基因的表达调控参与ESCC 的演进和放化疗抵抗机制。
综上所述,慢性低浓度镉暴露可诱导ESCC 细胞相关lncRNA 和mRNA 的表达谱发生改变,其可能通过复杂的lncRNA-mRNA 网络引起系列生物学过程、细胞组分、代谢与免疫、转录调控、信号转导的变化,从而构成镉促ESCC 发生发展和放化疗抵抗的复杂分子机制。本研究结果亦充分支持了本课题组前期研究工作揭示的镉暴露食管癌细胞株较亲本细胞具有更强的侵袭迁移能力、EMT和干细胞特性以及对放化疗的耐受性[13]。此外,本研究鉴定的若干关键lncRNA和mRNA 有望成为镉暴露ESCC 患者预后判断的潜在生物标志物和治疗靶点。
猜你喜欢
新民周刊(2022年27期)2022-08-01
传染病信息(2021年6期)2021-02-12
昆明医科大学学报(2020年12期)2021-01-26
山东医药(2017年20期)2017-07-01
中国学术期刊文摘(2016年1期)2016-02-13
西南军医(2016年1期)2016-01-23
中国继续医学教育(2015年4期)2016-01-07
川北医学院学报(2015年5期)2015-12-05
生物医学工程学进展(2015年1期)2015-02-28
山东医药(2015年44期)2015-02-28
