
氧化应激对骨髓间充质干细胞影响机制的研究进展*
2023-09-01 08:19俞卫娟高静媛综述冯林杰田发明审校
重庆医学 2023年16期
俞卫娟,高静媛 综述,冯林杰,田发明 审校
(1.华北理工大学附属医院全科医学科,河北唐山 063000;2.中国人民解放军联勤保障部队第982医院创伤科,河北唐山 063000;3.华北理工大学公共卫生学院,河北唐山 063000)
骨髓间充质干细胞(bone marrow stromal cells,BMSCs)是一种来源于骨髓的细胞,具有多向分化能力,可向骨、软骨、肌、成脂等谱系进行细胞分化[1-2]。BMSCs来源可靠,易于从骨髓中分离和扩增,便于获得免疫原性,是组织工程领域理想的种子细胞[3-4]。在多种组织损伤的动物模型中,BMSCs均显示出修复和重建受损组织更快、更好的再生能力,已广泛用于多种疾病的治疗,例如联合基因治疗肿瘤、脊髓损伤移植、自身免疫性疾病、神经系统相关疾病等,同时更是骨组织工程相关领域中的种子细胞[5-6]。骨稳态的维持取决于成骨谱系细胞和破骨谱系细胞之间的协调代谢活动,可使骨形成和骨吸收处于一种动态平衡[7-8]。当骨稳态失衡时,便会产生成骨不全症、骨质疏松症、骨折延迟愈合等代谢性骨病[9-10]。其中基本多细胞单位(basic multicelluler unit,BMU)作为调控骨稳态的细胞群,包括不同分化时期的成骨谱系细胞和破骨谱系细胞,而BMSCs作为成骨谱系细胞中的起源细胞,在上述疾病发生、发展中发挥了更为重要的作用[11-12]。活性氧(reactive oxygen species,ROS)在病理状态下诱导机体产生氧化应激反应,导致细胞内代谢紊乱,损伤蛋白质、脂质、DNA,使BMSCs增殖、分化受到抑制,凋亡增加,引起骨形成下降,致使骨代谢异常,骨稳态失衡[13-15]。本文通过汇总近年发表相关文献,针对氧化应激对BMSCs功能的影响及相关机制予以简要综述,有助于为骨代谢疾病提供潜在的治疗新思路。
1 氧化应激
氧化应激是指有机体在受到强烈的热刺激、机械刺激、化学刺激等有害刺激时,引起ROS产生过多,使体内抗氧化系统的防御和修复系统功能出现超负荷,导致氧化-抗氧化系统失去平衡,同时机体更倾向于氧化状态,进而对机体产生的一系列病理性氧化损伤[16]。丙二醛(malondialdehyde,MDA)是由氧化应激产生的氧化产物,可以作为过氧化指标,反映细胞氧化损伤程度的标志。抗氧化酶包括谷胱甘肽过氧化物酶(glutathione peroxidase,GPx)、超氧化物歧化酶(superoxide dismutase,SOD)、过氧化氢酶(catalase,CAT)、血红素加氧化酶-1(heme oxygenase-1,HO-1)等,具有清除自由基的能力,发挥抗氧化功能。
低水平的ROS可作为细胞内生理信号分子,为细胞传递信息,调控细胞的增殖与分化。而异常高水平的ROS则会增加线粒体膜通透性、线粒体肿胀、线粒体DNA损伤。这导致线粒体电子传递链功能障碍、三羧酸循环紊乱、ATP合成紊乱、跨膜电位降低、细胞色素C释放、凋亡因子激活、依赖性凋亡通路形成,抑制BMSCs的增殖与分化,最终诱导BMSCs凋亡[17]。氧化应激反应引起BMSCs内DNA、脂质过氧化及信号传导、转录的改变,以及基因表达变化从而使增殖能力降低,抑制分化,促进细胞凋亡[18-19]。研究发现,氧化应激通过调控相关细胞信号通路,影响内核基因的表达,抑制BMSCs的增殖、分化,诱导细胞凋亡,导致骨形成下降、骨量丢失和微观结构退变,进而出现一系列骨代谢疾病[20]。
2 ROS对BMSCs功能的影响
2.1 对细胞增殖的影响
研究表明,低浓度过氧化氢预处理BMSCs可明显上调SOD和CAT水平,以增强抗氧化损伤能力,促进BMSCs增殖,减少细胞凋亡[21]。缺氧常可引起氧化应激,而当缺氧因子基因被沉默后,ROS水平下降,细胞内Ca2+和NO水平升高,BMSCs存活率上升,促进细胞增殖和迁移[22]。
相关研究表明,氧化应激可能通过抑制Nrf2/HO-1通路抑制BMSCs的增殖能力,降低细胞活性[23-24]。正常生理情况下,细胞质中的核因子NF-E2相关因子(nuclear-factor erythroid 2-related factor 2,Nrf2)作为关键的抗氧化应激因子与Kelch样ECH关联蛋白1(Kelch-1ike ECH- associated protein l,Keap1)蛋白结合并以非活性的状态存在于细胞质中,其通过靶向蛋白酶降解从而保持Nrf2的低转录活性。在氧化应激条件下,机体的防御机制开启,Nrf2-Keap1的相互作用以剂量依赖的方式进行解离,Nrf2与Keap1解离后从细胞质中转移到细胞核,与细胞中防御相关的因子抗氧化反应元件(antioxidant-response element,ARE)结合,调节抗氧化蛋白,使HO-1等抗氧化酶表达增多,缓解氧化应激对细胞的损伤程度,逆转氧化应激对BMSCs增殖的负性作用[25-27]。HO-1是一种催化血红素降解的诱导酶,具有抗炎、抗氧化应激和抗凋亡特性。新的证据显示,HO-1在维持骨稳态中至关重要,这使得HO-1成为骨质疏松治疗的潜在靶点[28]。既往研究结果表明,异补骨脂素(isopsoralen,IPRN)可以通过抑制氧化应激和细胞凋亡来减少卵巢切除术(OVX)诱导的小鼠骨质流失,并促进HO-1诱导BMSCs的成骨分化,有望作为雌激素替代剂和天然抗氧化剂用于绝经后骨质疏松的治疗[29]。CORM-3是一种携带一氧化物并复制其生物作用的新型化合物,在低剂量时是无毒、安全且现成的CO替代品,能改善OVX大鼠模型中的HO-1及其产物CO明显降低的影响,从而延缓OVX大鼠体内的骨丢失[30]。同时,该研究的体外实验表明,补充外源性CO通过激活Nrf2/HO-1信号通路逆转过氧化氢诱导的线粒体功能障碍,通过调节巨噬细胞极化抑制破骨细胞分化。该研究揭示了低剂量CO在骨质疏松症治疗中的作用机制和应用潜力。
此外,激活Nrf2的重要上游调控因子磷脂酰肌醇-3-激酶(phosphatidylinositol 3 kinase,PI3K)/蛋白激酶B(protein kinase B,AKT)也可以降低氧化应激对BMSCs的不利影响,增强其增殖、迁移能力[31]。由于PI3K/AKT通路在细胞代谢中的关键作用,包括葡萄糖摄取、糖酵解、脂质合成、核苷酸合成和蛋白质合成,对于BMSCs的增殖功能至关重要。研究表明,通过激活PI3K/AKT/Nrf2或PTEN/PI3K/AKT信号通路,可以保护BMSCs免受氧化应激的氧化损伤。硒代L-甲硫氨酸是一种ROS清除剂,可以将过氧亚硝酸盐还原为L-甲硫氨酸硒氧化物,然后通过谷胱甘肽维持的非酶促反应恢复到其初始状态[32]。研究表明,硒代L-甲硫氨酸通过下调磷酸酯酶-张力蛋白同源物(phosphatase and tensin homolog,PTEN)表达水平,而上调p-PI3K、p-AKT水平及β-catenin、Runx2表达水平,抑制过氧化氢对BMSCs活力的不利影响,进而提高BMSCs增殖率、成骨相关蛋白表达水平。目前,已有相关报道白杨素激活PI3K/AKT/Nrf2通路,保护BMSCs免受氧化应激的影响,从而促进糖尿病SD大鼠颅骨缺损的骨再生[33]。
氧化应激还可以通过调控NMNAT3-烟酰胺腺嘌呤二核苷酸(NAD+)-SIRT3等相关通路来调节线粒体功能,从而影响BMSCs的增殖。过多的ROS会下调沉默信息调控因子样蛋白3(silent information regulator 3,SIRT3)和上调乙酰化SOD2,使线粒体基质内超氧化物歧化减少,导致细胞线粒体功能障碍而使抗氧化能力下降,抑制BMSCs的分化与增殖[34]。CHEN等[35]研究发现,白藜芦醇以剂量依赖的方式激活AMPK/PGC-1α/SIRT3轴增强SIRT3表达,明显减轻糖皮质激素诱导的股骨头坏死(glucocorticoid-induced osteonecrosis of the femoral head,GIONFH)模型大鼠氧化应激损伤,恢复BMSCs成骨潜能,防止GIONFH的发生。此外,JIANG等[36]研究结果显示,白藜芦醇还可以增强OVX小鼠对氧化应激的抵抗力,并通过激活SIRT1和FoxO1去乙酰以调节包括SOD在内下游靶点抗氧化酶的氧化损伤,加速BMSCs的成骨作用,可作为治疗骨质疏松症的新药理靶点。
此外,相关研究表明氧化应激也可能通过NMNAT3-NAD+-SIRT3轴上调SIRT3依赖的去乙酰化相关蛋白异柠檬酸脱氢-2(isocitrate dehychogenase 2,Idh2)和叉头转录因子O3a(forkhead box O3a,FoxO3a)乙酰化水平,从而对线粒体结构和功能的损伤加重,导致BMSCs活力下降,增殖能力减弱,阻滞细胞生长[37-38]。烟酰胺单核苷酸腺苷转移酶3(nicotinamide mononucleotide adenylyl transferase 3,NMNAT3)除了在合成NAD+中发挥重要作用,在氧化应激下条件下,过表达NMNAT3的BMSCs细胞内NAD+上调,SIRT3的活性增强,明显改变线粒体中Idh2和FoxO3a表达,有效改善氧化应激下BMSCs的线粒体结构,增加线粒体膜电位,增加ATP合成,促进线粒体生物发生调节因子过氧化物酶体增殖物激活受体γ共激活因子1α(peroxisome proliferator-activatedreceptorγcoactivator 1α,PGC-1α)和核呼吸因子-1(nuclear respiratory factor,NRF1)表达[39],见图1。

图1 氧化应激通过相关通路对BMSCs增殖的影响示意图
2.2 对细胞分化的影响
BMSCs是一种多分化的干细胞,其在整个发育过程中一直保持干性,直到被应激源和生理需求触发分化[40]。但是BMSCs分化为一种谱系后逐渐丧失向另一种谱系分化的潜能,即BMSCs定向成骨细胞分化后会阻止BMSCs向其他细胞分化[41]。BMSCs的成脂倾向是骨质疏松中海绵样骨形成的主要原因[42]。氧化应激不仅降低了BMSCs活力和增殖指数,而且还明显抑制BMSCs成骨方向的分化,反而促进其向成肌、成脂等其他方向分化,使成骨基因(包括ALP、OPN和Runx2)下调,导致骨形成减少,不利于成骨[43-46]。
氧化应激可通过调控无翅整合基因(wingless int,Wnt)/β-catenin通路抑制BMSCs成骨分化。经典Wnt信号通路由胞外Wnt配体通过卷曲蛋白(Fr2)家族中的7次跨膜受体和低密度脂蛋白受体相关蛋白5/6(Lrp5/6)传递信号,使β-catenin向细胞核聚集和易位,并与转录因子T细胞因子/淋巴细胞增强因子(T-cell factor/Lymphocyte enhancer factor,TCF/LEF)在细胞核相结合,从而调控下游通路靶基因Runt相关转录因子2(Runx2)和过氧化物酶体增殖物激活受体γ(peroxisome proliferator activated receptorγ,PPARγ)的表达,而这两个基因分别是成骨和脂肪形成的主转录因子[46-48]。正常情况下,Wnt/β-catenin信号通路在BMSCs定向成骨分化中发挥至关的重要作用,使BMSCs更趋向于成骨细胞分化[49]。研究表明,ROS可能通过激活c-Jun氨基末端激酶(c-Jun N-terminal kinases,JNK)信号通路然后直接磷酸化TCF,导致β-catenin与TCF结合减少,下调成骨相关因子表达,抑制BMSCs成骨分化,使BMSCs更趋向于成脂和成软骨方向分化[50]。相关研究发现,氧化应激可能通过抑制BMP/Smad通路降低SOD和GPx的活性,下调Runx2、ColⅠ、OCN等成骨相关基因表达,从而负性调节BMSCs的成骨分化,使其倾向于其他方向分化[51]。
转化生长因子-β(transforming growth factor-β,TGF-β)超家族成员中的骨形成蛋白-2(bone morphogenetic protein-2,BMP-2),在成骨分化中发挥重要作用的。氧化应激反应会抑制BMP的表达,使BMP-2与其受体(BMPR-Ⅰ、BMPR-Ⅱ)之间在细胞表面形成复合物减少,下调BMPR I对Smad信号的磷酸化表达;同时,TGF-β1与BMPR-Ⅱ的结合减少,抑制Smad下游通路激活,使得Smad与Runx2之间相互作用减少,最终引发炎症级联反应抑制抗氧化酶的活性,减少BMSCs的成骨分化[52]。有研究显示,桃叶珊瑚苷(aucubin,AU)不仅可以通过激活Nrf2/HO1信号通路,保护骨折愈合免受氧化应激损伤,还可以部分通过BMP2/Smads信号通路促进BMSCs成骨,促进骨折愈合[53]。
氧化应激也可以通过PI3K/AKT/FoxO1通路负向调控BMSCs向成骨谱系的分化[54]。过量ROS引起的氧化应激会激活FoxO信号通路发挥抗氧化功能,同时降低PI3K和AKT的磷酸化水平,进而使ALP活性、Runx2和β-catenin表达水平下降,导致BMSCs的成骨分化能力下降[55]。此外,过量的ROS会促进肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)的表达,形成一个正反馈回路,即一个恶性循环,可以激活下游靶点NF-κB信号通路,上调NADPH氧化酶(包括NOX1和NOX2)的表达,下调SOD1和CAT的表达[56]。
此外,氧化应激会阻断细胞外信号调节激酶(extracellular signal-regulated kinase,ERK)1/2信号通路,通过下调ERK和环磷腺苷效应元件结合蛋白(cAMP-response element binding protein,CREB)可以抑制成骨分化,上调半胱氨酸天冬氨酸蛋白酶3(caspase-3)激活、LDH水平,同时,PPARγ2和脂蛋白脂肪酶的表达增加,有利于成脂分化,抑制BMSCs成骨谱系分化,使骨形成降低[57-58],见图2。
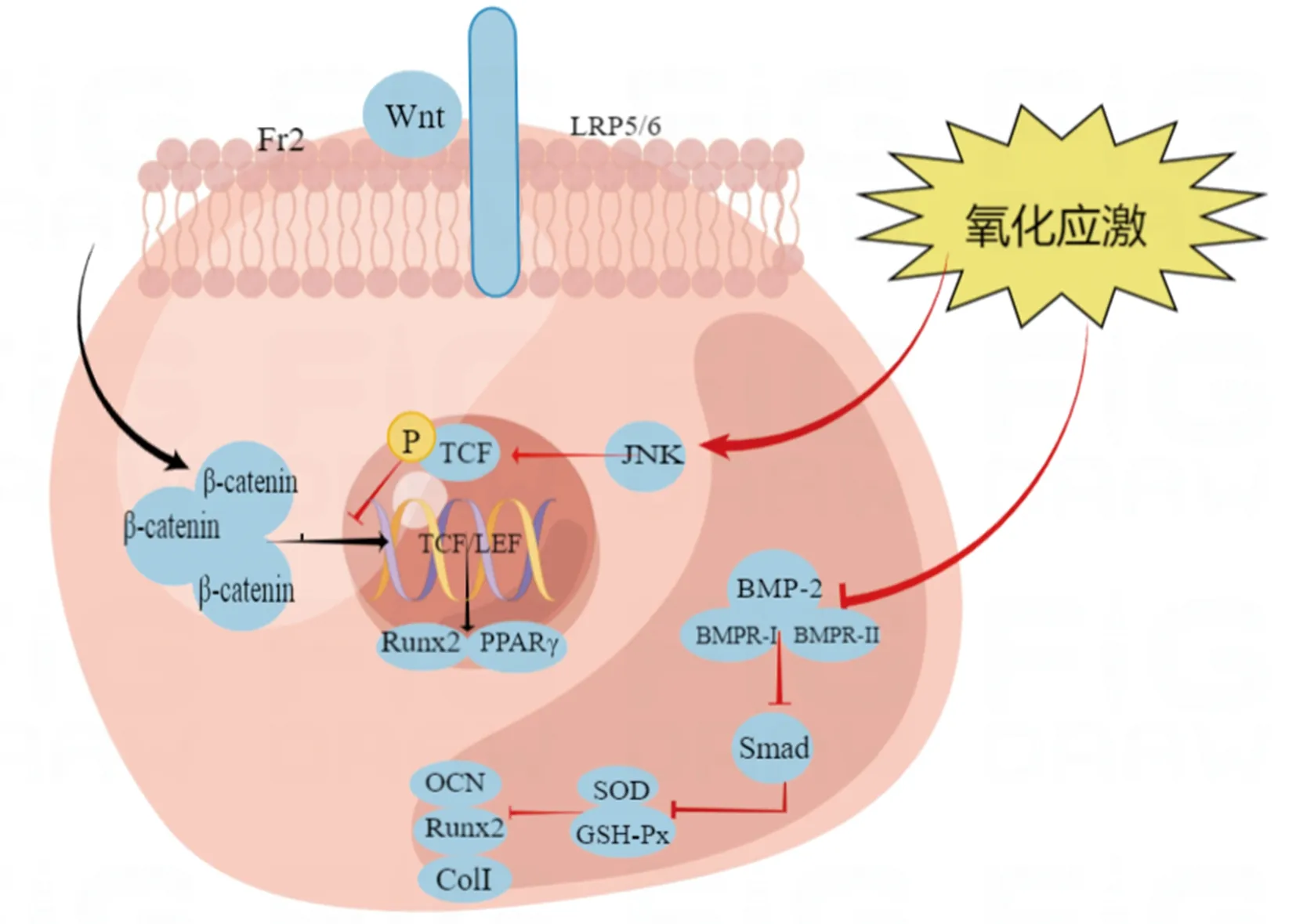
LRP:脂蛋白受体相关的蛋白5/6;OB:成骨细胞。图2 氧化应激通过相关通路对BMSCs分化的影响示意图
2.3 对细胞凋亡的影响
在氧化应激过程中,BMSCs的端粒长度缩短,细胞发生了复制性衰老,其增殖、分化能力均降低。氧化应激通过抑制关键的DNA修复因子(如53BP1),引起不可逆的细胞周期阻滞、细胞增殖抑制,而且衰老相关β-半乳糖苷酶的活性增加及激活p53、p21等细胞周期蛋白依赖激酶抑制因子表达增多,进而促进BMSCs的衰老过程,诱导BMSCs凋亡[59]。ROS水平的升高促进了JNK的磷酸化,该激酶从细胞质转移到线粒体,导致caspase-3激活,随后凋亡[60]。其次,在过氧化氢诱导的BMSCs模型中,氧化应激会上调促凋亡蛋白Bax、Bak和caspase-3的表达,同时下调抗凋亡蛋白B淋巴细胞瘤-2(B-cell lymphoma-2,Bcl-2)的表达,增强细胞色素C(cytochrome C,Cyt-c)释放,进一步激活caspase级联反应,促进caspase诱导的BMSCs凋亡[61-62]。此外,过多的电离辐射诱导机体内产生TNF-α,触发激活caspase-3和caspase-8;同时激活下游血清淀粉样蛋白A(serum amyloid A protein,SAA)等相关表达的细胞因子,并通过Toll样受体2(toll-like receptor 2,TLR2)和甲酰肽受体2(formyl peptide receptor 2,FPR2)两个受体来调节炎症反应因子,进而诱发炎症级联反应,触发ROS积累和持续的氧化应激,激活DNA损伤相关蛋白的表达,均可明显诱导BMSCs凋亡[63]。
ROS可诱导ERK、JNK、p38丝裂原活化蛋白激酶(p38 mitogen-activated protein kinases,p38MAPK)等丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)家族通路的激活,激活细胞凋亡因子、p53、p38MAPK和DNA损伤反应途径,诱导族BMSCs衰老和凋亡[64]。MARK家族是氧化应激最重要的下游信号通路之一,通常被认为是细胞存活、增殖和凋亡的调节因子。ERK通路是MAPK通路中的一个重要家族,主要由3个核心蛋白激酶(RAF、MEK和ERK)组成,相互间顺序激活,共同组成一个准确高效的信号传递网络,发挥其调节作用。帕金森病蛋白7(parkinson disease protein 7,PARK7)作为氧清除剂、抗氧化应激蛋白,在BMSCs存活中起着重要作用。过表达PARK7可上调BMSCs中MEK1/2、ERK1/2和E26转录因子1(ELK1)的磷酸化水平,增加ERK1/2核易位和SOD表达水平,促进BMSCs抗氧化应激过程,有效降低ROS和MDA水平,保护线粒体膜电位,改善过氧化氢诱导的BMSCs凋亡[65],见图3。
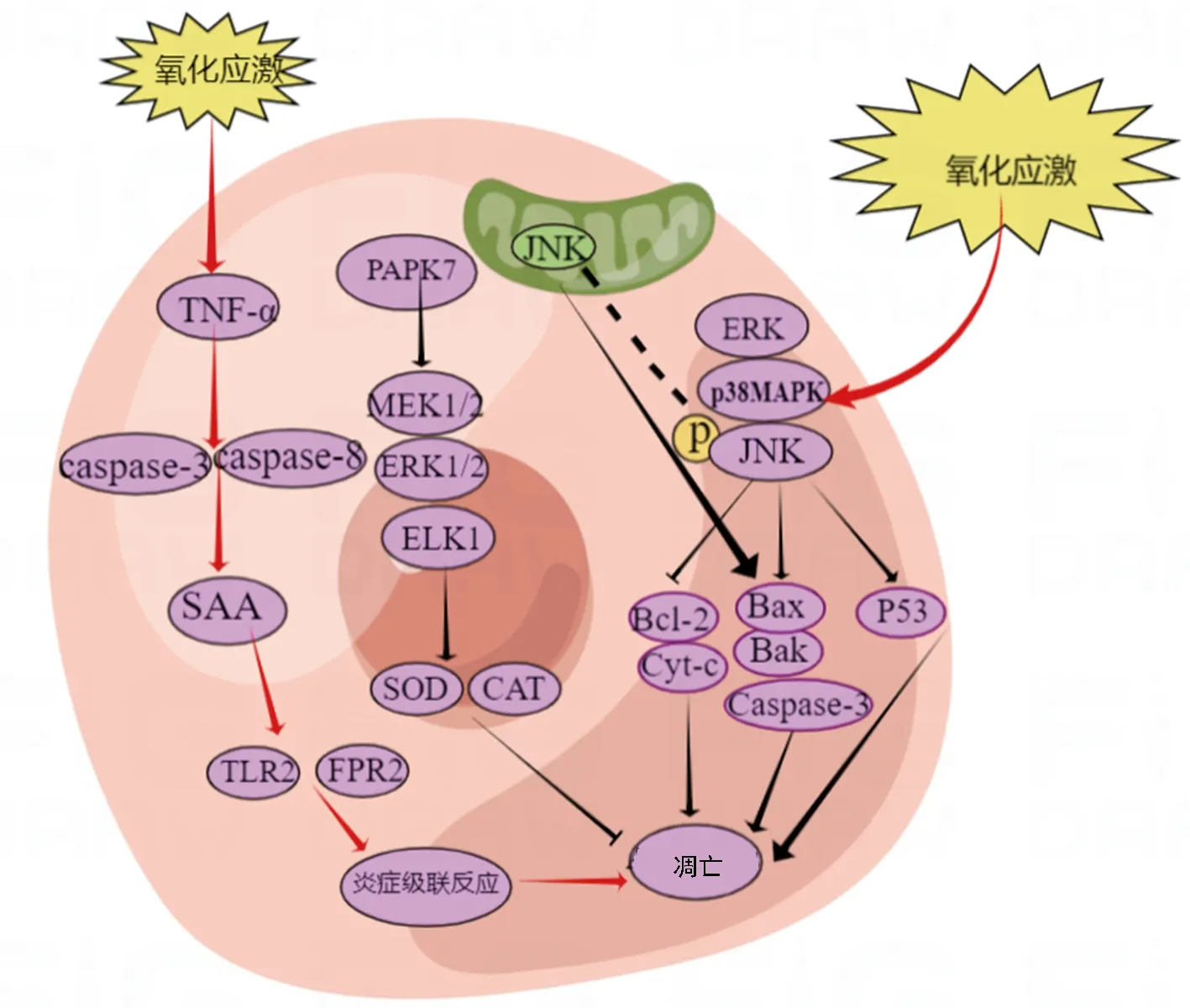
P53:衰老相关因子;PAPK7:帕金森病蛋白7。图3 氧化应激通过相关通路对BMSCs凋亡的影响示意图
3 小结与展望
综上所述,氧化应激已经成为骨代谢疾病中重要的研究领域之一,其通过对多种信号因子及信号通路的调控来抑制骨形成生理过程中BMSCs的增殖和分化,促进细胞衰老、凋亡,导致骨形成能力下降及骨稳态失衡,最终造成骨代谢异常。目前,已有部分研究以Nrf2/HO1、ERK1/2、PI3K/AKT/Nrf2及SIRT3、FoxO1等相关信号通路相关因子或竞争性抑制MAPK家族信号通路为靶点抑制氧化应激和炎症反应,同时促进BMSCs的成骨分化、成骨细胞的生成、血管生成、抗氧化应激、脂肪细胞凋亡和破骨细胞凋亡等积极的影响,协调骨代谢,有望作为开发骨质疏松症、骨折等骨代谢疾病的潜在治疗药物新靶点[66]。深入了解氧化应激对BMSCs的影响及机制对于开发新的促进骨形成药物,乃至骨质疏松、骨折、骨缺损等临床干预具有重要意义,后续研究中应进一步明确氧化应激对BMSCs的分子机制和级联网络,并探索开发相关靶向干预或联合干预,为上述疾病的治疗提供新的思路。
猜你喜欢
口腔医学(2021年10期)2021-12-02
海洋通报(2021年1期)2021-07-23
生物学通报(2021年4期)2021-03-16
世界科学技术-中医药现代化(2020年2期)2020-07-25
中华老年口腔医学杂志(2016年2期)2017-01-15
西南军医(2016年6期)2016-01-23
中国病理生理杂志(2015年8期)2015-12-21
天津护理(2015年4期)2015-11-10
西南军医(2015年2期)2015-01-22
癌变·畸变·突变(2014年1期)2014-03-01
