
BSCL2基因突变致先天性全身脂肪营养不良3例病例报告
2023-10-12 03:43苏雪莹李秀珍李翠玲郑锐丹梅慧芬盛慧英江敏妍邵咏贤
中国循证儿科杂志 2023年4期
苏雪莹 李秀珍 李翠玲 郑锐丹 梅慧芬 盛慧英 江敏妍 邵咏贤 张 文 刘 丽
1 病例资料
例1:男,1岁,G1P1,足月剖宫产,出生体重3.2 kg、身长47 cm。1月龄时发现全身脂肪缺失,头发浓密。3月龄出现倒三角脸,颈部、腋窝色素沉着,四肢及背部体毛增多增粗(图1A和C),体格检查和超声结果均显示肝大,实验室检查显示肝功能异常(AST 73 U·L-1,ALT 116U·L-1),血清TG升高(22.2 mmol·L-1),空腹胰岛素升高(115 mIU·L-1),多次查空腹血糖值偏高(15.7~21.0 mmol·L-1)。1岁时超声检查提示肝大(肝右叶斜径118 mm)、脾大(脾长径95 mm)。
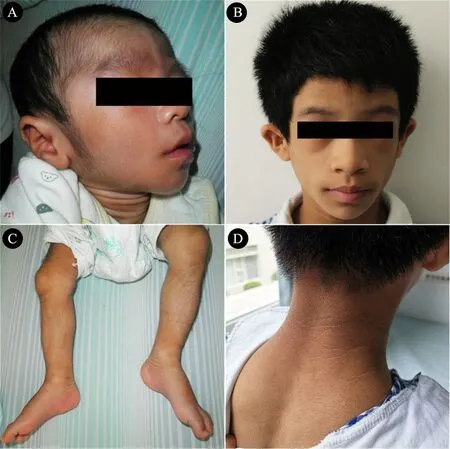
图1 BSCL2基因突变致先天性全身脂肪营养不良患儿的体貌特征
例2:女,3岁,G1P1,足月顺产,出生体重3.3 kg、身长48 cm。3月龄时发现全身脂肪菲薄,四肢肌肉明显。5月龄时当地医院检查发现血清TG升高(7.5 mmol·L-1),腹部超声提示肝大、脂肪肝。6月龄时体格检查发现患儿呈倒三角脸,毛发浓密,四肢肌肉发达,全身皮肤色素沉着;心电图大致正常;超声心动图显示房间隔缺损 (继发孔型),左室壁非对称性增厚并左室流出道梗阻;腹部超声提示肝大、脾大。2岁时发现生长发育缓慢,走路不稳。患儿目前3岁,脸颊凹陷呈倒三角形、毛发旺盛、颈部黑棘皮症(图1B和D),轻度智力障碍和语言发育迟缓,超声显示肝大 (肝右叶斜径127 mm)。
例3:例2胞弟,2月龄,G2P2,足月顺产,出生体重3.1 kg、身长48 cm。生后1周纳奶差,全身皮下脂肪明显减少,血清TG升高(8.0 mmol·L-1)。1月龄时超声检查提示肝大、脂肪肝。2月龄时体检发现患儿脸颊凹陷,全身毛发增多,四肢肌肉强壮。
在3例患儿诊断中分别取得其父母的知情同意后,抽取患儿及其父母的静脉血各2 mL,提取基因组DNA,运用Sanger测序技术进行BSCL2(NM_032667.6)基因突变分析,将PCR产物纯化后送至华大基因公司行双向测序,将测得的序列与参考序列比较,确定致病基因突变。3例均检测到BSCL2基因复合杂合变异,例1携带c.757G>T (p.Glu253*)和c.975insG (p.Ile326Aspfs*12),例2和例3均携带c.565G>T (p.Glu189*)和c.782dupG (p.Ile262Hisfs),均为HGMD报道的已知致病性变异。患儿父母均为单杂合变异携带者。
3例均有全身脂肪缺失、多毛、肌肉发达、肝大等先天性全身脂肪营养不良(CGL)的典型临床特征,结合基因检测结果,均确诊为2型CGL(CGL2)。例1和例2由母乳过渡到辅食后采取低脂饮食,在保障生长发育的营养基础上,合理限制高热量食物的摄入,饮食控制半年后,2例患儿TG水平较初诊时下降约50%。例1在3月龄时出现空腹血糖、胰岛素水平升高,确诊糖尿病[1,2],予胰岛素皮下注射(每天2 U·kg-1),1个月后血糖显著下降,胰岛素剂量减少至每天1 U·kg-1,血糖可维持在正常范围。例3至本文随访截止时尚不足6月龄,仍以母乳喂养为主,末次随访TG为22.1 mmol·L-1。随访过程中,3例均未出现肾衰竭、进行性中枢神经系统损伤等严重并发症。
2 文献复习
文献检索时间为1990年1月至2022年12月,以“先天性全身脂肪营养不良”和“BSCL2”为关键词,检索中国知网、万方数据知识服务平台和维普资讯中文期刊服务平台;以“congenital generalized lipodystrophy”、“BSCL2”和“Chinese”为关键词检索PubMed 数据库。
共检索到中文文献10篇[3-12],英文文献11篇[13-23],共计纳入43例中国的BSCL2基因突变相关CGL患者。男25例,女18例,中位确诊年龄为3个月(12 d至28岁),其中1例为成年确诊,46.5%(20/43)在1岁内确诊。临床表现:均在出生半年内出现全身脂肪缺失,27例(63%)出现黑棘皮症,35例(81%)合并全身多毛,39例(91%)出现肝大,25例(58%)合并脾大,14例(33%)并发糖尿病,8例(19%)出现肥厚性心肌病, 20例(46%)合并智力低下,4例(9%)合并严重进展性中枢神经系统损伤。基因突变情况:21例 (48%)携带复合杂合突变,52%为纯合突变;最常见的突变位点为c.782dupG(37%,16/43),携带该变异的16例患者中12例为纯合突变,4例为复合杂合突变;其他主要突变位点为c.565G>T 7例(16%)、c.974dupG 5例(12%),其余突变位点所占比例均低于10%,为散发低频突变。
3 讨论
CGL是一种罕见的常染色体隐性遗传病,主要表现为全身脂肪组织缺失,可伴有高TG血症、肝大、黑棘皮症、糖尿病以及肥厚性心肌病等[24]。按照致病基因的不同,CGL分为1 ~ 4型,分别由AGPAT2、BSCL2、CAV1和PTRF基因突变所致[25]。目前全球报道的CGL患者约500例,中国报道的CGL患者43例且约95%为BSCL2基因变异导致的CGL2型[22]。
BSCL2基因定位于染色体11q13,编码的seipin蛋白为内质网膜蛋白,有两个跨膜结构域[26],主要表达于脂肪组织、中枢神经系统和睾丸精子细胞。研究证实seipin在TG的聚集、脂滴生成以及脂滴形态的维持中发挥重要作用[27, 28]。Seipin缺失影响脂肪细胞的分化[29, 30],导致脂肪组织的大量缺失及脂肪营养不良。脂肪组织的大量缺失导致其分泌的瘦素和脂联素水平显著降低,引起糖耐量受损及胰岛素抵抗等代谢异常状态[31]。此外,脂肪组织缺失使得大量的TG、甘油二酯异位蓄积于肝脏,肝脏脂肪变性又进一步促进胰岛素抵抗的发展,引起血糖升高[32]。由于皮下和内脏脂肪组织的广泛缺失,CGL2往往比其他3型更严重,患者除有CGL常见临床症状外,还可伴发肾衰竭、智力障碍、肥厚性心肌病以及严重中枢神经系统损伤[33-36]。

目前我国CGL患者治疗原则以对症治疗为主。CGL常见的并发症为高TG、脂肪肝和高血糖,因此饮食控制非常重要。本文例1和例2由母乳转为添加辅食后遵循低脂饮食的原则,TG水平明显下降,而月龄较小的例3为纯母乳喂养,在随访的6个月内其TG水平持续升高。国内外研究均证实低脂饮食可有效改善患者代谢情况,对于儿童来说,要优先保证其生长发育的需要,采用配比合理的健康饮食[25]。对于部分严重高脂血症且饮食控制效果不佳的患儿,可加用贝特类药物。对于伴有糖尿病的患者,可根据实际情况口服二甲双胍、磺酰脲类、噻唑烷二酮类药物或注射胰岛素控制血糖。多国的临床研究表明,美曲普汀可长期有效改善CGL患者的高TG血症、高血糖和高胰岛素血症,并可改善性激素紊乱等内分泌代谢异常[44-46]。但由于价格昂贵,且长期用药的有效性和安全性仍有待验证,因此尚未获得中国食品药品监督管理局(CFDA)批准上市。
本研究经广州市妇女儿童医疗中心伦理委员会批准 (伦理批号:2014-1-49) 。
猜你喜欢
英语世界(2023年6期)2023-06-30
中老年保健(2021年9期)2021-08-24
昆明医科大学学报(2021年8期)2021-08-13
种子(2021年3期)2021-04-12
中国生殖健康(2020年2期)2021-01-18
小学生导刊(2018年13期)2018-06-29
外语教学理论与实践(2016年1期)2016-06-11
河南医学研究(2014年5期)2014-02-27
河南医学研究(2014年2期)2014-02-27
华东理工大学学报(自然科学版)(2014年1期)2014-02-27
