
Recent advances in annulations enabled by nucleophilic Lewis base/metal dual catalysis☆
2023-11-18 09:53QinWngYinggoMengLuluWuErQingLi
Chinese Chemical Letters 2023年10期
Qin Wng, Yinggo Meng,*, Lulu Wu, Er-Qing Li,*
a College of Chemistry, Green Catalysis Center, Zhengzhou University, Zhengzhou 450001, China
b School of Science, Henan Agricultural University, Zhengzhou 450002, China
Keywords:Lewis base Metal catalysis Annualtion Dual catalysis Synthetic methods
ABSTRACT Metal/nucleophilic Lewis base dual catalysis has been recognized as a reliable and promising strategy for finishing ideal organic synthesis over the past decades.The new strategy can usually achieve some chemical reactions that cannot be realized by the traditionally mono-catalytic system, dramatically expanding the synthetic utility of chemical transformations by leveraging additional activation modes.Thus considerable progress has been made in the synthesis of a wide range of heterocyclic and biologically active compounds by using the combination of diversely metal/nucleophilic Lewis base dual catalysts, including metal/phosphine, metal/N-heterocyclic carbene (NHC) and metal/tertiary amine dual catalysis systems.In this review, we describe a comprehensive and updated advance of metal/nucleophilic Lewis base dual catalytic annualtion reactions, meanwhile, the related mechanism and the application of these annulation strategies in natural product total synthesis will be highlighted in detail.
1.Introduction
Dual catalysis is a powerful and reliable synthetic strategy for the construction of a wide range of pharmaceuticals and bioactive natural products.In dual catalysis, two different catalysts activate diverse reactive substrates concurrently by either cooperative or relay catalytic process in a single chemical transformation.Thus it can realize unprecedented chemical transformations that could not be achieved by either of the catalysts alone.In the past decades, numerous progress on dual catalysis involving metal/metal, metal/organocatalyst and organo/organocatalyst has attracted considerable attention.In particular, the combination of transition metal with organocatalysis has attracted interest from a large number of research groups since the proof of concept was reported in 2001 [1,2].And a large number of unprecedented annualtion reactions involving transition metal with organocatalyst dual catalysis have been developed in laboratories worldwide [3–10].
As a class of organocatalysts, nucleophilic Lewis bases, mainly including tertiary phosphine,N-heterocyclic carbene (NHC) and tertiary amine catalysts, have emerged as promising and versatile catalysts over the last few decades.For each reported nucleophilic Lewis base-catalyzed reaction, the reactive substrates are limited to electron-deficient olefins, alkynes, aldehydes, ketones and imines.Nevertheless, the electron-rich substrates, which are commonly used in metal catalysis, often suffer from significant limitations, including slow conversion, poor selectivity, or even no reaction at all.In recent years, by mimicking the characteristic of biosystems, the strategy of metal/Lewis base dual catalysis has been explored by organic chemists for achieving important transformations that are unobtainable through single-catalyst systems [11–14].In such systems, metal catalysts are widely used in bond-breaking and bondforming events, and nucleophilic Lewis base catalysts exhibit a excellent tolerance of functionalities with a unique level of regio- and stereocontrol.Thus two distinct catalysts can work either cooperatively or independently to fulfill ideal organic synthesis in one operation, dramatically reducing solvents, waste, time,etc.
The utilization of dual catalysts in a single reaction to realize correspondingly chemical synthesis has witnessed continuous success.In mechanism, dual catalysis was divided into two types including cooperative dual catalysis and relay or sequential catalysis.Dual cooperative catalysis describes the concept of two different catalysts working together synergistically without interfering with one another but rather by simultaneously activating two different reactive species, one an electrophileAand the other a nucleophileB.In relay or sequential catalysis, the intermediateA-Bwas first obtained in the presence of catalyst 1, and then intermediateABwas transformed into the desired productCunder catalyst 2(Scheme 1).In dual catalytic system, some more reactive intermediates and sophisticated transformations may produce, the key to the realization of dual catalysis system is thus that each catalyst precisely activate the corresponding substrates.In other words, The compatibility problems is critical for the success of dual transition metal/nucleophilic Lewis base catalytic system.For this reason, the combination of diverse catalysts for cooperative or relay activation requires a combination of strategy and empiricism in evaluating compatible systems for the ultimate goal of exploring new reaction modes.

Scheme 1.Metal/nucleophilic Lewis base dual catalysis.
Given the power and efficiency of dual transition metal/nucleophilic Lewis base catalytic annualtion reactions in constructing valuable ring structures, and their remarkable applications in pharmaceuticals and bioactive natural products synthesis.Enormous efforts have been devoted to the development of highly efficient protocols for the synthesis of this motifsviadual transition metal/Lewis base catalytic cycloaddition reactions in the past decade.Thus a comprehensive and update review focusing on recent advancements in transition metal/nucleophilic Lewis base catalytic system-empowered annulation reactions and its applications in the total synthesis of natural products is essential.In this review, we summarize and classify the related research works based on different catalytic pathways such as transition metal/phosphines, transition metal/amines and transition metal/NHC dual catalysis.We sincerely hope that this tutorial review will act as a handy reference for synthetic chemists interested in exploring and discovering more types of multiple catalytic reactions.
2.Application of phosphine/metal dual catalytic system in annulation reaction
2.1. Phosphine/metal cooperatively catalytic annulations
Owning a pair of nonbonding electron pairs, tertiary phosphines are not only applied as ligands that coordinate with the metal center, but also can be directly used as organocatalysts to catalyze the chemical reactions [15–20].That is to say, in the dual catalytic system, tertiary phosphines can coordinate with the transition metals involved, which will result in the deactivation of catalytic system, thereby diminishing or even preventing the individual reactivity of each substrate.Thus rare examples have been reported involving transition metal/phosphine dual catalytic system.In 2003, Krische and coworkers were the first to resolve the compatibility problems of palladium complex and tertiary phosphines,realizing the intramolecular enone cycloallylation reactions by applying Pd(PPh3)4and PBu3cooperatively catalytic system.In this report, the authors used allylic carbonates as latent “nonclassical” electrophiles, achieving the precise activation of nucleophilic(enone) and electrophilic (allyl carbonate) partners through phosphine addition and metallo-π-allyllation, respectively.In mechanism, the authors thought that the lifetime of the transiently generated enolate would be extended through solvation in the form of hydrogen-bond interactions, in which capture of the metalloπallyl intermediate would be facilitated (Scheme 2) [21].

Scheme 2.Phosphine/palladium cooperative catalyzed cycloallylation.

Scheme 3.Totally synthesis of (±)-quinine and (±)-7-hydroxyquinine.
Using the phosphine/palladium cooperative catalytic system,Krische and coworkers realized concise stereoselective synthesis of (±)-7-hydroxyquinine and (±)-quinine.As shown in Scheme 3,(±)-7-hydroxyquinine8awas obtained in 13 steps and 11% overall yield from aminoacetaldehyde diethyl acetal.And (±)-quinine8bwas synthesized in 16 steps and 4% overall yield from commercial aminoacetaldehyde diethyl acetal (Scheme 3) [22].
Despite those efforts, phosphine/palladium cooperative catalytic system has not seen widespread use up to date, This mainly unsolved challenge in the design of such a cooperative system has been the difficulty of controlling compatibility problems.Until 2019, Chen and coworkers reported an auto-tandem cooperative catalysis for Morita–Baylis–Hillman carbonates from isatins and allylic carbonates using a simple Pd(PPh3)4precursor, affoding a spectrum of spirooxindoles incorporating a 4-methylene-2-cyclopentene motif in good yields with chemoselectivity [23].In mechanism, the authors thought that dissociated phosphine from Pd(PPh3)4generated phosphorus ylides12and the Pd concurrently led toπ-allylpalladium complexes13.And they underwent aγregioselective allylic-allylic alkylation reaction (14), followed by a cascade intramolecular Heck-type coupling to produce the desired spirooxindoles11.It was worth noted that the authors first realized the phosphine/palladium cooperative catalyzed asymmetric cycloaddition reaction, obtaining high yield with moderateeevalue(Scheme 4).
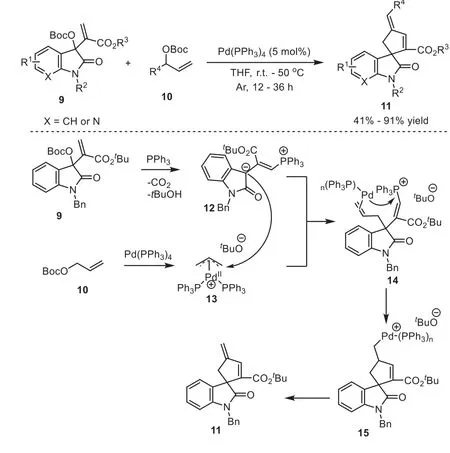
Scheme 4.Auto-tandem cooperatively catalysis for Morita–Baylis–Hillman carbonates from isatins and allylic carbonates.

Scheme 5.Phosphine/palladium cooperatively catalytic [3+4] annulation reaction of Morita-Baylis-Hillman carbonates and vinyl benzoxazinanones.

Scheme 6.A possible mechanism for phosphine/palladium cooperative catalytic[3+4] annulation reaction.
Azepine derivatives are abundant in many natural products,bioactive molecules, and pharmaceuticals.Thus, the development of an efficient catalytic system for the construction of these molecules is still needed.In 2019, Li and co-workers developed a phosphine/palladium cooperatively catalytic [3+4] annulation reaction of Morita-Baylis-Hillman carbonates and vinyl benzoxazinanones, providing a range of vinyl 2,3-dihydro-1Hbenzo[b]azepine derivatives in moderate to good yields and diastereoselectivities (Scheme 5) [24].
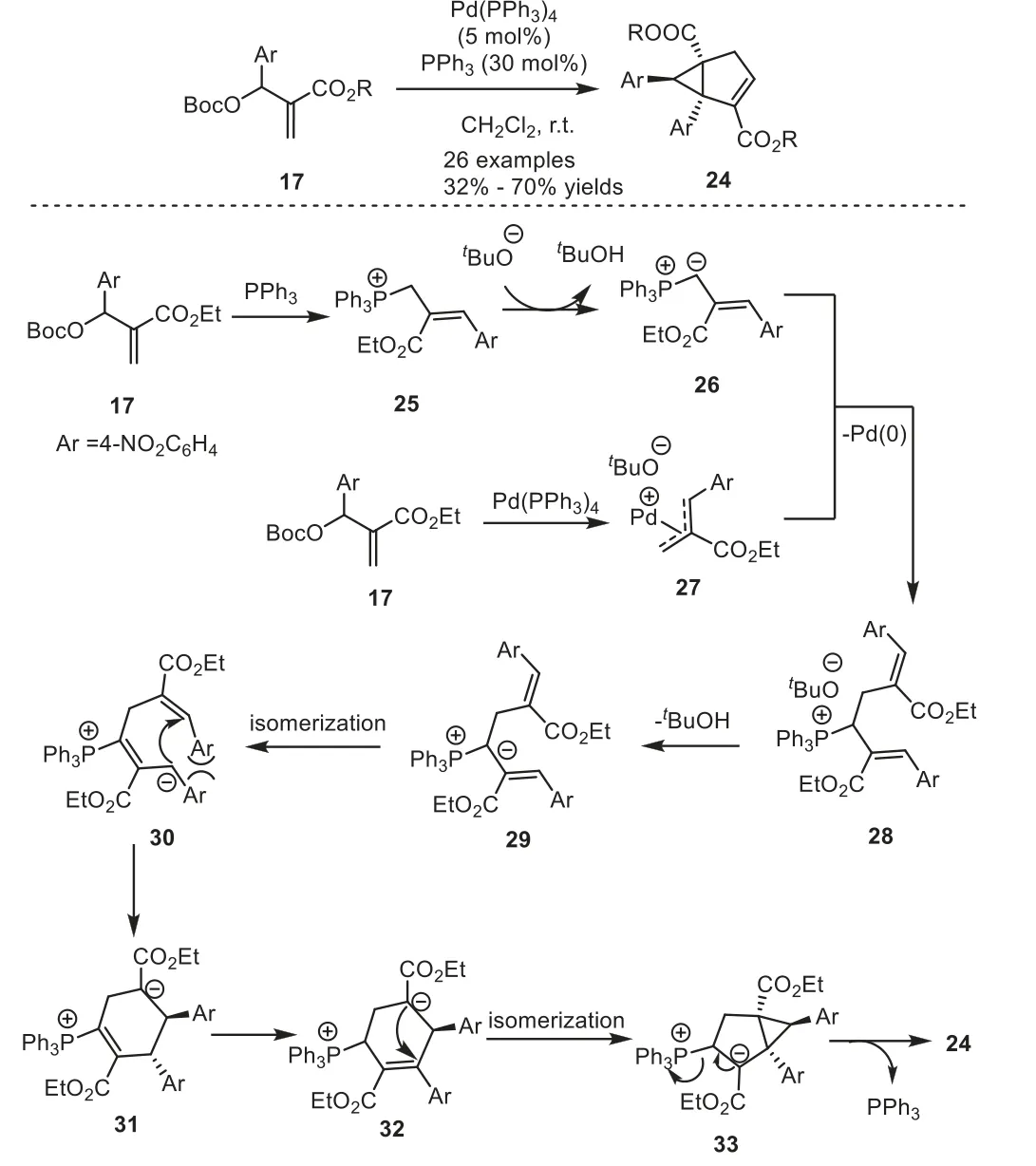
Scheme 7.A possible mechanism for phosphine/palladium cooperative catalytic successive [2+3]/[2+1] cycloadditions.
A plausible mechanism is presented, as outlined in Scheme 6.The reaction was first triggered by nucleophilic attack of PPh3on MBH carbonates17to yield zwitterion19, Simultaneously,16and Pd(PPh3)4produced theπ-allylpalladium complex20.Next, theγregioselective addition of the zwitterionic intermediate19to theπ-allylpalladium complex20gave the intermediate21and regenerated Pd(0) catalyst.An intramolecular Michael addition occurred to produce the intermediate22.Then the electron-transfer process and subsequentβ-elimination of PPh3afforded the corresponding intermediate23.Finally, two-step isomerization of C=C bond produced the observed adducts18in the presence of base (Scheme 6).
In 2021, Li and coworkers reported a phosphine/palladium cooperatively catalytic successive [2+3]/[2+1] cycloadditions of MBH carbonates, leading to the desired bicyclo[3.1.0]hexenes24in moderate to good yields [25].For the mechanism, the authors found that one molecule of MBH carbonates with PPh3produced a tertiary phosphine-tethered allylic P-ylide26.Simultaneously, another molecule of MBH carbonates coordinated with Pd(PPh3)4to obtainπ-allylpalladium complex27.Subsequently, anα-regioselective allylic alkylation (28), dehydrogenation (29), proton shift (30), ring-closure produced intermediate31.Finally, a sequential isomerization (32), annulation (33) and regeneration of the PPh3catalyst led to the desired adducts24(Scheme 7).
2.2. Phosphine/metal sequentially catalytic annulations
In dual catalysis, sequential catalysis (or tandem/cascade catalysis) is a very powerful and efficient approach in organic synthesis.In this field, the two catalysts are operating in series; the first catalyst activates the substrates toward the formation of the reactive intermediate, which is then transformed in the next catalytic cycle in the presence of the second catalyst, and leads to the desired product [26].The key challenge in sequential catalysis is to aviod deactivation of two catalysts by interaction.In 2009, Wu and coworkers first resolved the compatibility problems and reported a silver triflate and triphenylphosphine sequential catalyzed domino reaction of the three-component reaction of 2-alkynylbenzaldehyde, amine, andα,β-unsaturated ketone [27].In 2012, Kwon and coworkers developed a phosphine/palladium sequentially catalytic annulation reaction.Notably, the reaction underwent sequential phosphine-catalyzed nucleophilic addition followed by Pd(PPh3)4-catalyzed Heck cyclization.In addition, the authors accomplished concise total syntheses of 3-deoxyisoochacinic acid (37), isoochracinic acid (38), and isoochracinol (39) (Scheme 8) [28].

Scheme 8.Phosphine/palladium relay catalytic cycloaddition reaction.

Scheme 9.Phosphine/palladium sequential catalytic Michael-Heck annulation of 2-iodobenzylmalonates.
In 2015, Kwon and coworkers used 2-iodobenzylmalonates and methyl propiolate as reactive substrates to test the viability of Michael-Heck annulation in forming carbocycles.Under phosphine/palladium sequential catalysis, the tandem Michael-Heck annulation proceeded smoothly, affording the desired alkylidene indane41in 38%-99% yields with moderateZ/Eselectivity [29].The experiment result suggested that the salt additives had a significant effect on the yield, and an excellent yield was achieved when using tetra-n-butylammonium acetate as the additive.It was worth noted that alkylidene indane could be translated into a nonsteroidal anti-inflammatory drug (Sulindac)43from known literature (Scheme 9) [30,31].
3.Application of NHC/metal dual catalytic system in annulations
3.1. NHC/metal cooperatively catalytic annulations
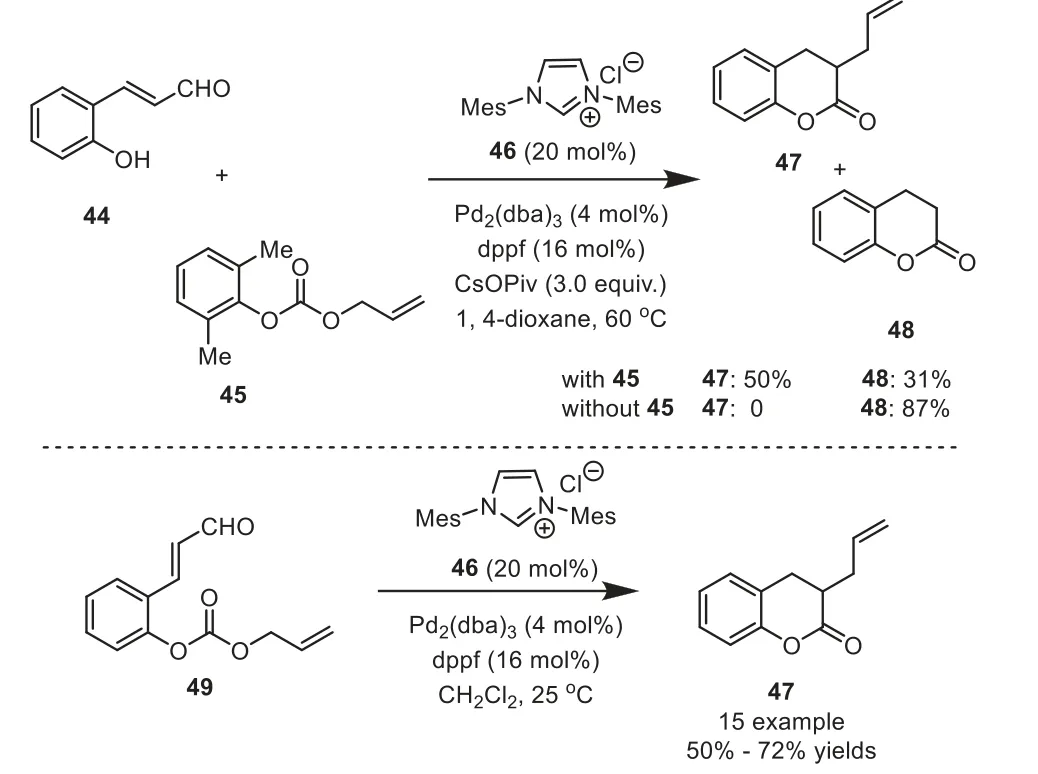
Scheme 10.Cooperative NHC/palladium catalysis approach to access a variety of 3-allyl dihydrocoumarin derivatives.
N-Heterocyclic carbene (NHC) catalysis is recognized as a powerful tool for the construction of complex organic frameworks from simple and readily available starting materials [32–38].However,NHC catalysis is typically limited to established substrate classes and reaction modes.Thus cooperative combination of transitionmetal catalysis and NHC catalysis could dramatically expand the scope of NHC catalysis and provide access to currently inaccessible reaction pathways.In 2014, Scheidt and coworkers reported a new cooperative NHC/palladium catalytic approach to access a variety of 3-allyl dihydrocoumarin derivatives.Initially, the authors investigated the intermolecular reaction of44with allyl carbonate45using a cooperative NHC-Pd catalysis system.Obviously, the undesired dihydrocoumarin48was obtained as a competing by-product[39].The authors hypothesized that the phenolic proton promoted the tautomerization of the NHC-enolate to the NHC-acyl adduct.To overcome this limitation, they designed a new substrate (o-alloc aldehyde49) to mask the phenol with the allyl source.Thus the desired allylated dihydrocoumarins47were produced in 50%-72%yields (Scheme 10).
In 2016, Glorius and coworkers developed an asymmetric cooperative system for the intermolecular [4+3] cycloaddition reaction that combines transition-metal/NHC organocatalytic system [40].This asymmetric process was first to demonstrate the compatibility of these two important catalytic modes and efficiently afforded annulated 1-benzazepine products with excellent regio- and enantioselectivities.A plausible “cooperative activation” concept was proposed in this paper: First, the coordination of vinyl benzoxazinanone50to the palladium catalyst formed an electrophilic allylpalladium(II) complex54upon decarboxylation, simultaneously,the nucleophilic addition of NHC organocatalyst to the enal51gave rise to the NHC-homoenolate55.Then, the NHC-homoenolate55underwent conjugate addition to the allyl-palladium(II) complex,affording the acyl azolium56and released the palladium catalyst.Finally, this species underwentN-acylation cyclization to furnish the final product53and regenerated the NHC organocatalyst52(Scheme 11).

Scheme 11.The intermolecular [4+3] cycloaddition reaction that combines Pd/NHC cooperative system and plausible mechanism.

Scheme 12.The intermolecular [2+5] cycloaddition reaction that combines NHC/Pd cooperative system.
Later, Glorius and coworkers reported the detailed mechanistic studies involving a palladium catalyst and an NHC organocatalyst dual catalytic systems.This result revealed that the NHC, besides its role as an organocatalyst, also fortuitously acts as a ligand on the active metal catalyst [41].In 2018, Glorius and coworkers expected to report an enantioselective [2+5] annulations involving such NHC/transition metal cooperativity to generate challengingε-caprolactones in a highly enantioselective fashion [42].Unfortunately, no product was detected when the authors used NHC precatalyst58and monodentate PPh3, the authors thought that NHC58could easily compete with the weakly coordinating PPh3for ligation to Pd, which inhibited the reaction.In this context, the authors questioned whether the ligation of NHC to the transition metal was prevented and the NHC was only operative in the organocatalytic cycle, which would thus provide access to currently inaccessible reaction pathways by allowing the fine-tuning of both catalytic systems.Based on this strategy, some more strongly chelating bidentate ligands were used to optimize the reaction condition.When a more bulky and electron-rich ligand (R)-Tol-BINAP was used, the desired product60was obtained in 76% yield with excellentee(>99%ee).Further mechanistic studies showed that use of a bidentate phosphine ligand played key role in preventing the binding of NHC to the transition metal and thereby the success of this transformation (Scheme 12).
In 2020, Du and coworkers used 3-substituted but–2-enoates and 1-tosyl-2-vinylaziridine as reactive substrates, developing a cooperative NHC/palladium-catalyzed [2+3] annulation system [43].In this dual catalytic system, the NHC catalyst and the palladium catalyst could work well independently instead of quenching each other, affording the desired (E)-3-ethylidene-4-vinylpyrrolidin-2-one skeletons in medium to good yields (Scheme 13).

Scheme 13.Cooperative NHC/palladium [2+3] annulation of 3-substituted but–2-enoates and 1-tosyl-2-vinylaziridine.

Scheme 14.RuCl3/NHC dual cooperative catalytic [3+3] cycloaddition.
Besides Pd/NHC cooperatively catalytic system, Huang and coworkers reported an enantioselective [3+3] cycloaddition reaction by RuCl3/NHC catalytic system [44].In this reaction, theα,βunsaturated acylazolium intermediate reacted selectively with 1,3-dicarbonyl compounds or ketones at either theβ- orγ-carbon,producing polysubstituted chiral lactones in high yield and with excellent enantioselectivity (up to 98% yield, 94%ee).The authors thought that the key oxidation step in this reaction was a Rumediated SET process.In order to determine the nature of the radical, the radical clock experiments were carried out, the result showed the relative configurations of the cyclopropanes were perfectly preserved during both the oxidation and the annulation,which suggested the radical was strictly localized at the carbonyl carbon, with little resonance to the correspondingβ-radical isomer.According to this, a plausible mechanism was proposed as shown in Scheme 14.Initially, the enal reacted with the NHC to yield a homoenolate55, RuCl3then oxidized the homoenolate by an SET process.The resulting radicalcation was deprotonated to give a zwitterionic radical species.Consequently, a second SET by Ru led to theα,β-unsaturated acylazolium, which undergoes[3+3] annulation with the 1,3-dicarbonyl compound (Scheme 14).
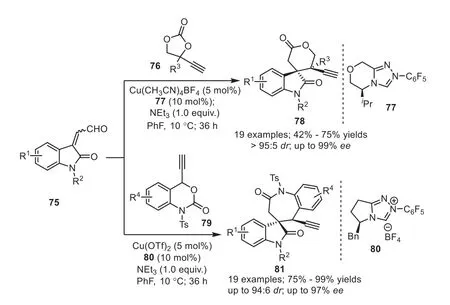
Scheme 15.NHC/copper cooperatively catalyzed [3+3] and [3+4] annulations between isatin-derived enals.
Copper-catalyzed asymmetric decarboxylation of ethynylethylene carbonates and ethynyl benzoxazinanones that generates nucleophile/copper-allenylidene bifunctional intermediates, provides a powerful approach to build chiral heterocycles.Inspired by this, cooperative dual catalytic system involving copper catalyst has received comprehensive interest.In 2019, Gong and coworkers reported a NHC/copper cooperatively catalyzed [3+3] and [3+4]annulations between isatin-derived enals75with ethynylethylene carbonates76and ethynyl benzoxazinanones79, affording the chiral spirooxindole derivatives with high structural diversity and enantiopurity (Scheme 15) [45].The experiment result showed that a chiral copper/NHC complex might form and participate in the control of stereochemistry, together with the chiral NHC catalyst.
Thus the authors proposed a plausible reaction mechanism.Initially, the active species82was formed by substrate76coordinating with a Cu(I)/NHC complex.Then sequential deprotonation/decarboxylation of82gave a copper-allenylidene intermediate (84or84′).Simultaneously, the Breslow intermediate85′ was produced by the addition of NHC organocatalyst to the enal75,which would thereafter react with copper-allenylidene intermediate84to generate an intermediate86.Finally,O-acylation cyclization and protonation furnished the final product78and regenerated the NHC organocatalyst and the copper catalyst (Scheme 16).
In 2020, Ye and Zhang utilized the copper/N-heterocyclic carbene dual catalytic system, developing a cooperatively catalyzed[3+4] annulation of salicylaldehydes with aziridines [46].The reaction proceeded smoothly under the optimized reaction conditions, producing the desired 1,4-benzoxazepinones in good yields with exclusive regioselectivity.Further study showed that the addition of NHC to the salicylaldehyde afforded the Breslow intermediate, which played a predominant role in improving the regioselectivity (Scheme 17).

Scheme 16.A possible mechanism for NHC/copper cooperatively catalyzed [3+3]and [3+4] annulations between isatin-derived enals.
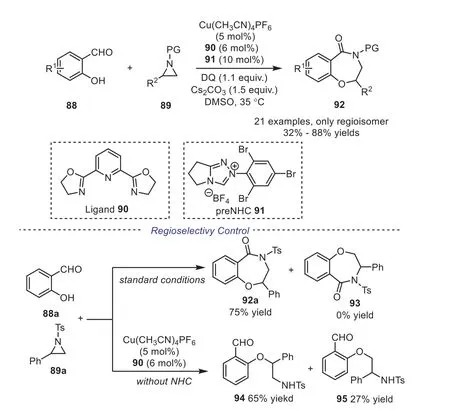
Scheme 17.Copper/NHC cooperatively catalyzed [3+4] annulation of salicylaldehydes with aziridines.
Catalytic kinetic resolution and dynamic kinetic asymmetric transformation of racemic mixtures is of great significance in synthetic chemistry.In 2021, Gong and Song reported a highly effi-cient chiral copper/NHC cooperatively catalytic systems for kinetically controlled processes, producing enantioenriched spirooxindolyl lactams in good yields with stereoselectivity by asymmetric[3+3] annulation [47].The experiment result showed that NHC not only was applied as organocatalyst in the reaction, but also played the key role in improving the catalytic activity of the copper complex as an additional ligand.It was worthy noted that kinetic resolution and dynamic kinetic asymmetric reaction could be switched by changing the loading of NHC, respectively.Namely,the use of 2.5 mol% Cu(CH3CN)4PF6, 5 mol% diphosphine96and 5 mol% NHC97was confirmed as the best optimized catalyst system for the catalytic kinetic resolution, while the presence of 5 mol%Cu(CH3CN)4PF6, 10 mol% diphosphine96and 2.5 mol% NHC97was found as the best dual system for the dynamic kinetic asymmetric transformation (Scheme 18).More recently, the authors realized an asymmetric [3+3] cycloaddition of racemic vinyl epoxides with isatin-derived enals, providing straightforward access to highly enantioenriched 3,3′-disubstituted oxindoles in good yields with good diastereoselectivity and excellent enantioselectivity [48].
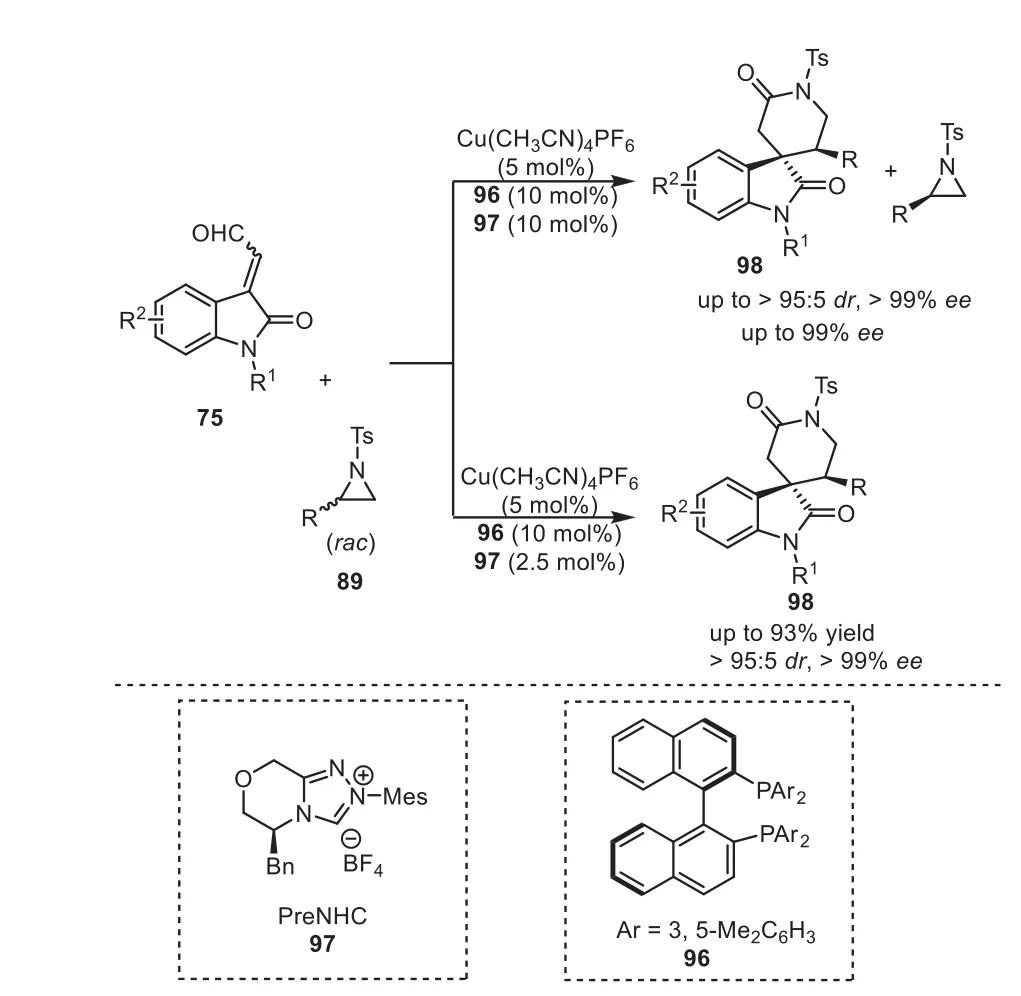
Scheme 18.NHC/copper cooperatively catalyzed [3+3] cycloaddition of aziridines.
γ-Butyrolactones display important structural skeletons that widely occur in natural molecules, pharmaceuticals, and functional materials.Therefore, highly efficient access to and fully characterizing all possible stereoisomers ofγ-butyrolactones are still needed.In 2020, Glorius and coworkers reported a stereodivergent NHC/Ir dual catalytic [2+3] cycloaddition reaction, providing all four stereoisomers ofα,β-disubstitutedγ-butyrolactones in good yields with excellent enantioselectivity by switching the four possible combinations of NHC precatalyst101(Cl) with the P–olefin ligand102.In addition, the obtained product was applied for total synthesis of the naturally occurring lignan (-)-hinokinin, which displayed anti-inflammatory, antimicrobial and modulatory effects on human GABA (γ-aminobutyric acid) transporter activities (Scheme 19) [49].
In 2015, Glorius and coworkers reported a switchable asymmetric annulation reaction with aromatic azomethine imines, in which homoenolate and enolate intermediates derived from enals could be switched under the catalysis of NHC in a rational and predictable manner [50].In 2021, Deng and coworkers applied this strategy to realize the stereoselective and regiodivergent reactions of enalsviacooperative NHC/iridium catalysis system [51].According to Glorius’s reports, an effective proton concentration would determine conversions between homoenolate and enolate forms.Thus the authors first used Et3N (100 mol%) as a base in the presence of the iridium complex [Ir]-A andN-heterocyclic carbene in CH2Cl2at room temperature, the [2+3] cycloaddition reaction could proceed smoothlyviaenolate intermediate, producing pyrrolo[1,2-a]indoles in good yields with regio-, diastereoand enantioselectivities.When the strong organic base DBU (100 mol%) was used in PhCF3, The homoenolate-derived products pyridine[1,2-a]indoles were obtained with complete regioselectivities.Similar to Glorius’s reports, all four stereoisomers of these products could be afforded by the pairwise combination of two chiral catalysts.In mechanism, the authors found that the ligand exchange did not occur when the NHC was used with the Ir complex ligated in the reactive system, which was different from Glorius’s reaction system (Scheme 20).
3.2. NHC/metal relay catalytic annulations
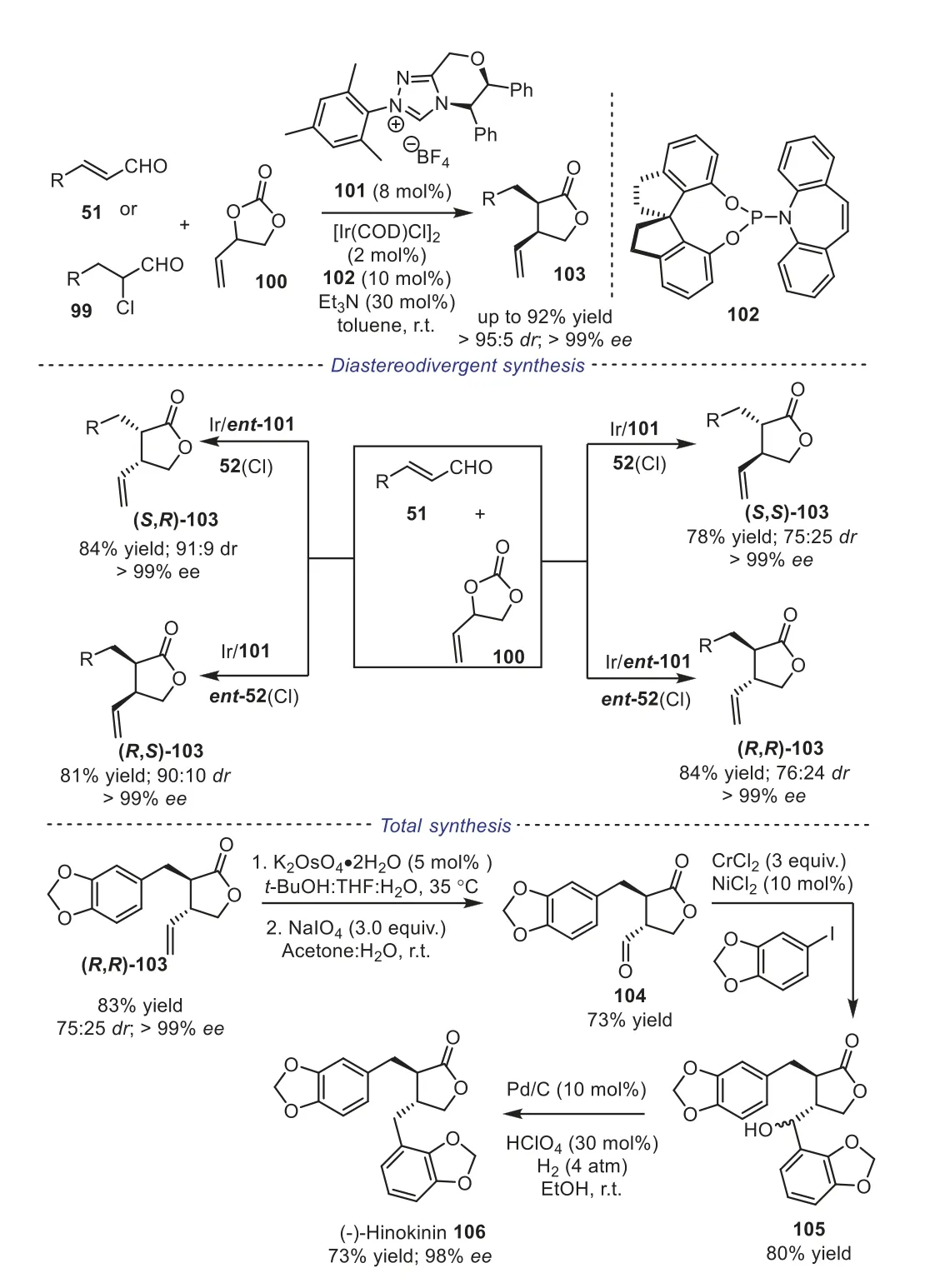
Scheme 19.NHC/Ir dual catalytic [2+3] cycloaddition of trans-cinnamaldehyde and vinylethylene carbonate.

Scheme 20.NHC/iridium catalysis enables stereoselective and regiodivergent[2+3] and [3+3] annulation reactions.

Scheme 21.Palladium/NHC relay catalytic domino reaction.
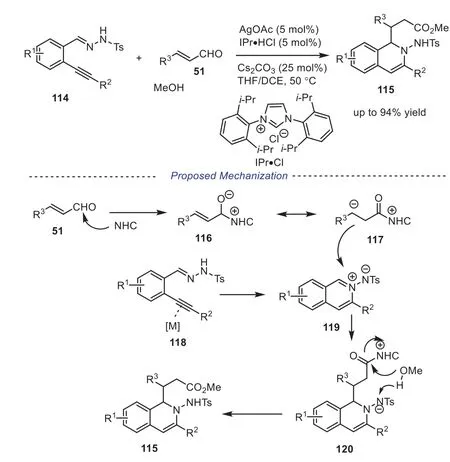
Scheme 22.Ag/NHC relay catalytic three-component reaction of N’-(2-alkynylbenzylidene)-hydrazide, methanol with α,β-unsaturated aldehyde.
Besides cooperatively dual catalytic system, the dual catalysis involving relay catalysis by means of the combination of an NHC organocatalyst and a transition-metal catalyst has also emerged as a powerful strategy for the development of new reactions.In 2006,Hamada and coworkers reported a palladium/NHC relay catalytic domino reaction including palladium-catalyzed allylic amination-NHC-catalyzed Stetter reaction cascade, affording the 3-substituted 2,3-dihydroquinolin-4-ones113in high yields [52].The authors found that the reaction rate was increased in the presence of 1.0 equiv.of acetic acid, that is to say, ammonium salt (AcOHPr2NEt) would function as Brønsted acid and accelerate the reaction (Scheme 21).
N′-(2-Alkynylbenzylidene)hydrazide is usually used as substrate in the Ag-catalyzed 6-endo-cyclization, producing the isoquinolinium-2-yl amide which is a good electrophile [53–55].Thus the 1,2-dihydroisoquinoline core can be applied in dual relay catalytic cycloaddition.In 2010, Wu and coworkers disclosed Ag/NHC relay catalytic three-component reaction ofN′-(2-alkynylbenzylidene)-hydrazide, methanol withα,β-unsaturated aldehyde, producing the 2-amino-1,2-dihydroisoquinolines115in good yield [56].In mechanization, the isoquinolinium-2-yl amide119was first obtained by Ag-catalyzed 6-endo-cyclization ofN′-(2-alkynylbenzylidene)hydrazide.Next, the Breslow intermediate117was obtained under the catalysis of NHC catalyst, which would attack the isoquinolinium-2-yl amide119to generate intermediate120.Finally, deprotonation of methanol and nucleophilic addition of carbonyl compound to produce the desired 2-amino-1, 2-dihydroisoquinoline115(Scheme 22).

Scheme 23.Ag/NHC relay cataltytic domino annulation reaction of N′-(2-alkynylbenzylidene)hydrazides with cyclopropane-carbaldehydes.
Under NHC catalysis, cyclopropanecarbaldehydes could act as anα-nucleophile or aγ-nucleophile in different reactions byinsituformation of enolate intermediates [57–59].In 2020, Cheng and coworkers reported a Ag/NHC relay cataltytic domino annulation reaction, providing an efficient approach for the construction of a variety of (1R,4R,5S)-4-(acylethyl)-11-aryl-2-tosyl-1,2,4,5-tetrahydro-5,1-(azenometheno)benzo[c]azepin-3-one122[60].In experiment, the reaction proceeded smoothly under catalysis of AgOCOCF3(10 mol%) to obtain the intermediate119, which then underwent an NHC-catalyzed annulation to produce the desired adducts122.Under respective optimized reaction conditions, the authors studied the substrate scope ofN′-(2-alkynylbenzylidene)hydrazides with cyclopropane-carbaldehydes,and the corresponding products122were obtained in moderate to good yields, with excellent enantioselectivity and diastereoselectivity.Based on the results of experiments, a possible mechanism was proposed for this transformation.First, theN-iminoisoquinolinium ylide intermediate119was formed by an Ag-catalyzed 6-endocyclization ofN′-(2-alkynylbenzylidene)-hydrazide, the enolate intermediate 125, on the other hand, was generatedin situfrom the cyclopropane-carbaldehydes under catalysis of NHC, which then attacked the C=N bond of isoquinolinium ylide119yielded the intermediate126.Finally, an intramolecular lactamization, following by 1,3-shift of the amide nitrogen producing the desired adduct122(Scheme 23).
In 2014, Chi and coworkers realized a Cu/NHC relay catalytic three-component domino reaction, the reaction involved a coppercatalyzed activation of alkynes and activation of ketenimine intermediates by anN-heterocyclic carbene organocatalyst.Studying the compatibility of Cu and NHC catalysts [61], the authors found that strong base promoted the coordination of NHC and copper compared with weak bases.That was say, under ‘weak’base conditions, there was a controllable kinetic/thermodynamic window that allowed the carbene organocatalyst and Cu metal catalyst to coexist with a meaningful level of concentrations.The experiment result confirmed this conclusion, when the authors used NHC/Cu catalyst in a ratio of 1:1 gave much better results (10 mol% each,79% yield) than using the preformed NHC–Cu complex (10% yield),which suggested that the two catalysts (NHC and Cu) independently (rather than as the metal-carbene complex form) work in the presence of weak bases.In addition, mechanism of the threecomponent domino reaction was illustrated in Scheme 24.Initially,Cu-catalyzed activation of alkyne to react with TsN3to obtain the ketenimine intermediate137, which was then activated by the NHC organocatalyst to form azolium enamide intermediate138.The intermediate reacted with reactive ketones and imines to form the desired product132, which was simultaneously transformed to alkene product133when the N-protected isatins were used as reactive substrates (Scheme 24).

Scheme 24.Cu/NHC catalyzed relay activation of alkynes for stereoselective reactions.

Scheme 25.NHC/Cu relay catalysis in the reaction of o-alkynylbenzaldehydes with N-acylimines.
1-Indanone scaffolds are widely present in agrochemicals and pharmaceuticals.In 2018, Cheng and coworkers disclosed an efficient approach for the construction of a pair ofZ- andE-2-amido-3-benzylidene-1-indanones in 47%-92% total yields by using NHC/Cu relay catalysis [62].Optimization of reaction conditions suggested that the reaction media and temperature played decisive roles on the cascade catalysis in question.In addition, this work also provided simple and efficient methods for the highly selective synthesis of multifunctionalized 1,3-dihydroisobenzofuran derivatives from the same reactants by using NHC/Cs2CO3relay catalytic system (Scheme 25).
Scheme 26.NHC/Pd cascade catalytic method for the synthesis of 2,2-disubstituted benzofuran-3-ones.

Scheme 27.Au/NHC relay catalytic cycloisomerization/cyclization reactions of ynamides and enals.
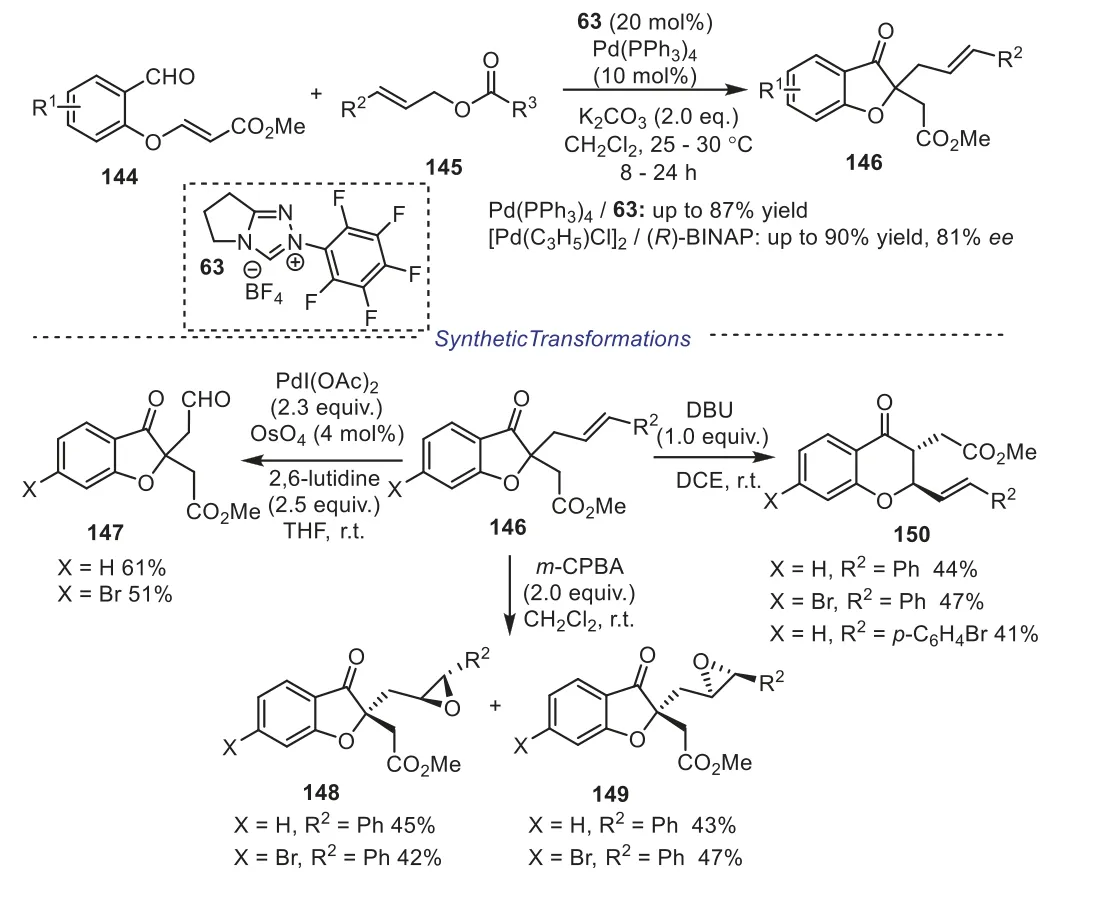
In the same year, Cheng and coworkers reported an effi-cient NHC/Pd relay catalytic method for the synthesis of 2,2-disubstituted benzofuran-3-ones in moderated to good yields, the reaction underwent a cascade Stetter reaction and regioselective allylation [63].When the reaction was performed in toluene at 0°C, using [Pd(C3H5)Cl]2/(R)-BINAP as the chiral palladium catalyst, the optically active 2-allylbenzofuran-3-one-2-acetates were unprecedentedly obtained in 54%-90% yields with 68%-81%ee, respectively.In addition, the obtained products could be easily converted into different polyfunctionalized compounds under suitable catalytic conditions, which demonstrated the valuable synthetic utility of the resulting products (Scheme 26).
In 2020, Chi and coworkers reported an Au/NHC relay catalytic enantioselective cycloisomerization/cyclization reactions of ynamides and enals, forming the bicyclic lactam products with excellent diastereo- and enantioselectivities [64].Control experiment showed that the use of AgPF6was essential to release the active gold catalyst from PPh3AuCl, while AgPF6alone did not have the ability to activate the alkyne substrate in the absence of PPh3AuCl.Thus the authors proposed a possible mechanism.As outlined in Scheme 27.Initially,α,β-unsaturatedN-sulfonyl ketimine156as a key intermediate was obtained by successive Au-catalyzed the activation of ynamide and isomerization.Next, treatment of enal with NHC formed Breslow intermedaite157that underwent a proton shift to give an azolium enolate intermediate158.Finally, the bicyclic lactam product154was afforded by azadiene Diels–Alder reactions and regenerated the both Au and NHC catalysts (Scheme 27).

Scheme 28.Au/oxidative NHC relay catalytic cascade annulation of α-aminoynones and enals.
Success in the NHC and transition metal dual catalytic system mainly focused on the cycloaddition of homoenolate/enolate intermediates with various dipoles possessing nucleophilic and electrophilic moieties generated under transition metal catalysis.More recently, Lu and coworkers reported an Au/oxidative NHC relay catalytic annulation ofα-amino-ynones and enals, in which pyrrolin-4-one (generatedin situfromα-amino-ynones under Au catalysis) served as a highly effective doubly nucleophilic synthon, affording pyrrole-fused lactones161in good to high yields with excellent enantioselectivities [65].The authors proposed a possible mechanism as showed in Scheme 28.First, enolate165was produced by cascade Au-catalyzed intramolecular cyclization ofαamino-ynones, protonolysis of intermediate164and deprotonation of pyrrolin-4-one.Concurrently, the Breslow intermediate55(producing by reaction of enal with NHC) underwent oxidation to form anα,β-unsaturated acyl intermediate166.Next, successively enolate conjugate addition and alkoxide cyclization occurred to form intermediate167, which finally underwent elimination to produce161with regenearation of the NHC catalyst (Scheme 28).
4.Application of tertiary amine/metal cooperatively catalytic system in annulations
4.1. Tertiary amine/metal cooperatively catalytic annulations
The tertiary amines own a pair of nonbonding electron pairs,which represent the most commonly recognized form of nucleophilic Lewis base catalysts [66,67].As the development of Lewis base catalysis, tertiary amine catalysts have proven to be effective catalysts for a range of synthetic transformations.In this review,we summarize recent progress in this area, and the diverse reactivities, various reaction modes and proposed mechanisms will be described in detail.
The Wynberg reaction reported in 1982 [68], has found widespread applications for the addition of ketene to di- and trichloroaldehydes and ketones.However, the Wynberg reaction involving substituted ketenes has been hampered by the tendency of these ketenes to dimerize under nucleophilic catalysis.In 2002,Lectka and coworkers reported a [2+2] cycloaddition reaction by using tertiary amine/In(OTf)3dual catalysis [69].According to experiment result, the authors thought that a chiral Lewis base was paired with an achiral Lewis acidic metal salt to effect a highyielding synthesis of optically enrichedβ-lactam products.Later,the authors validated the reaction mechanism by systematically mechanism experiment (Scheme 29) [70].

Scheme 29.Tertiary amine/In(OTf)3 cocatalyzed [2+2] cycloaddition of ketene with imine.

Scheme 30.Tertiary amine/Sc(OTf)3 co-catalyzed [2+2] cycloaddition reaction of substituted ketenes with unactivated arylaldehydes.
In 2005, Calter and coworkers described a tertiary amine/Sc(OTf)3co-catalyzed [2+2] cycloaddition reaction of substituted ketenes with unactivated arylaldehydes, affordingβlactones in high diastereo- and enantioselectivity [71].According to the experiment result, the authors thought that the diastereoselectivity of adducts depended on the substitution of the acid chloride.The reaction of aliphatic acid chlorides with aldehydes produced predominantly thetrans-isomers, and the substrates alkoxyacetyl chlorides favored formation of thecis-isomer (Scheme 30).
The reaction of copper complexes with propargylic esters generates copper–allenylidene complexes, which turn out to be versatile intermediates and allow the development of various propargylation reactions.In 2017, Gong and coworkers established a cooperative isothiourea/copper co-catalyzed asymmetric decarboxylative[2+4] annulation of 4-ethynyl dihydrobenzooxazinones and carboxylic acids, yielding optically active 3,4-dihydroquinolin-2-one derivatives in good yields with excellent stereoselectivities [72].Optimization of reaction suggested that the matched chirality of chiral ligand and organocatalyst played the key role in controlling diastereoselectivity.In addition, the enantioselectivity was predominantly determined by organocatalyst, even if the achiral copper complex, in combination with chiral isothiourea180, still obtained a high enantioselectivity.Subsequently, the substrate scope and limitation was investigated, and found that simple propargylic acetate or 3-phenylpropanoic acid was not suitable substrates in the reaction (Scheme 31).Almost simultaneously, Wu and coworkers reported a similar experiment result [73].

Scheme 31.Cooperative tertiary amine/copper co-catalyzed asymmetric decarboxylative [2+4] annulation of 4-ethynyl dihydrobenzooxazinones.

Scheme 32.A plausible mechanism for isothiourea/copper co-catalyzed asymmetric decarboxylative [2+4] annulation.
According to the results of experimental results and the previous reports, a plausible mechanism was presented.As shown in Scheme 32, the decarboxylation of79with a copper complex generated the copper–allenylidene intermediate182.Concurrently, a C1 ammonium enolate intermediate184was obtained by the treatment of a chiral isothiourea catalyst with an active electrophilic species183.The intermediate186was generated by an enantioselective nucleophilic addition of182to theγ-carbon atom of184.The authors thought that the copper complex [74] might play a dual role in which one copper participated in the decarboxylation of79to give the copper–allenylidene complex, and the other acted as a counterion bonded to the C1 ammoniumZ-enolate (185).Finally, a lactamization reaction of the intermediate186produced the 3,4-dihydroquinolin-2-one181and released the chiral isothiourea catalyst (Scheme 32).
Hydantoins as core structural elements have significant application potential in natural products and pharmaceuticals.In 2018,Gong and coworkers developed an isothiourea/copper cooperative catalytic strategy for the preparation of highly enantioenriched hydantoins by an enantioselectiveα-amination of esters [75].The reaction proceeded smoothly at 70°C in the presence of an isothiourea catalyst and a copper chloride-tributylphosphine complex by using CHCl3as a reaction solvent, affording the desired hydantoin190in good yield with enantioselectivity.It’s worth mentioning that this work provided simple and efficient methods for the highly efficient synthesis of an ester193, which could be transformed into (+)-CP-99994 (a high affinity NK1 antagonist) by following Shi’s procedure (Scheme 33) [76].

Scheme 33.Isothiourea/copper cooperative catalysis strategy for the preparation of highly enantioenriched hydantoins.

Scheme 34.A plausible mechanism for isothiourea/copper cooperative catalysis strategy for the preparation of highly enantioenriched hydantoins.
Based on the experimental results, a plausible mechanism of thisα-amination of esters was proposed as shown in Scheme 34.An acylation reaction of the ester187with a chiral isothiourea produced a chiral acylammonium197.Simultaneously, the fourmembered Cu(III) species195or a Cu(II) radical species196was obtained by the cleavage of the N-N bond of diaziridinone188in the presence of Cu(I) catalyst.The electron paramagnetic resonance (EPR) showed that the radical intermediate196might be the active copper species in the cooperative catalytic cycle.Next,the chiral intermediate199was produced by the treatment of the acylammonium197with Cu(II) species196.Subsequently, continuously reductive elimination of199and an intramolecular amidation reaction of200gave the enantioenriched hydantoin190and regenerated the catalyst (Scheme 34).
In 2021, Deng and coworkers reported an asymmetric decarboxylative [2+3] cycloaddition of ethynyl indoloxazolidones with carboxylic acids by using isothiourea/copper synergistic catalysis [77].In this reaction, a novel indolyl copper-allenylidene amphiphilic intermediates generatedviacopper-mediated decarboxylation of ethynyl indoloxazolidones and prepared a wide range of pyrrolo[1,2-a]indoles with excellent stereoselectivity.In addition,the resulting products could underwent click reaction and reduction reaction to give the desired 1,2,3-triazole and chiral indoline,respectively (Scheme 35).

Scheme 35.Isothiourea/copper synergistic catalyzed [2+3]-cycloaddition of ethynyl indoloxazolidones.
The Morita–Baylis–Hillman (MBH) adducts have emerged as powerful synthons in organic synthesis [78,79].In 2019, Chen and coworkers developed an efficient tertiary amine/iridium complexes cooperatively catalytic strategy for construction of valuable 4-azepane or 4-piperidine motifs by a [3+4] or [3+3] annulation pattern [80].It was worth noting that the chiral iridium complex induced the stereocontrol in this reaction, a common tertiary amine (DABCO), in combination with chiral iridium complex, could offer a high enantioselectivity, which differed from the known tertiary amine/metal catalysis.The substrate scopes and limitations of the cooperative catalytic strategy were also investigated under the optimized conditions.Besides carbamate-functionalized allylic carbonate207, the cyclic vinyl carbamate209and vinyl NTs aziridines212could be applied for the reactions of Morita–Baylis–Hillman carbonates by using tertiary amine/iridium complexes cooperative catalysis.Additionally, the obtained products could be further converted into more complex compounds under suitable catalytic conditions, which demonstrated the valuable synthetic utility of the product (Scheme 36).In 2021, the authors also realized asymmetric [3+4] annulations between isatinderived Morita-Baylis-Hillman carbonates and two types of vinyl carbonates by using synergistically tertiary amines and palladium catalytic system, affording a range of oxepane frameworks in moderate to good yields with high stereocontrol [81].
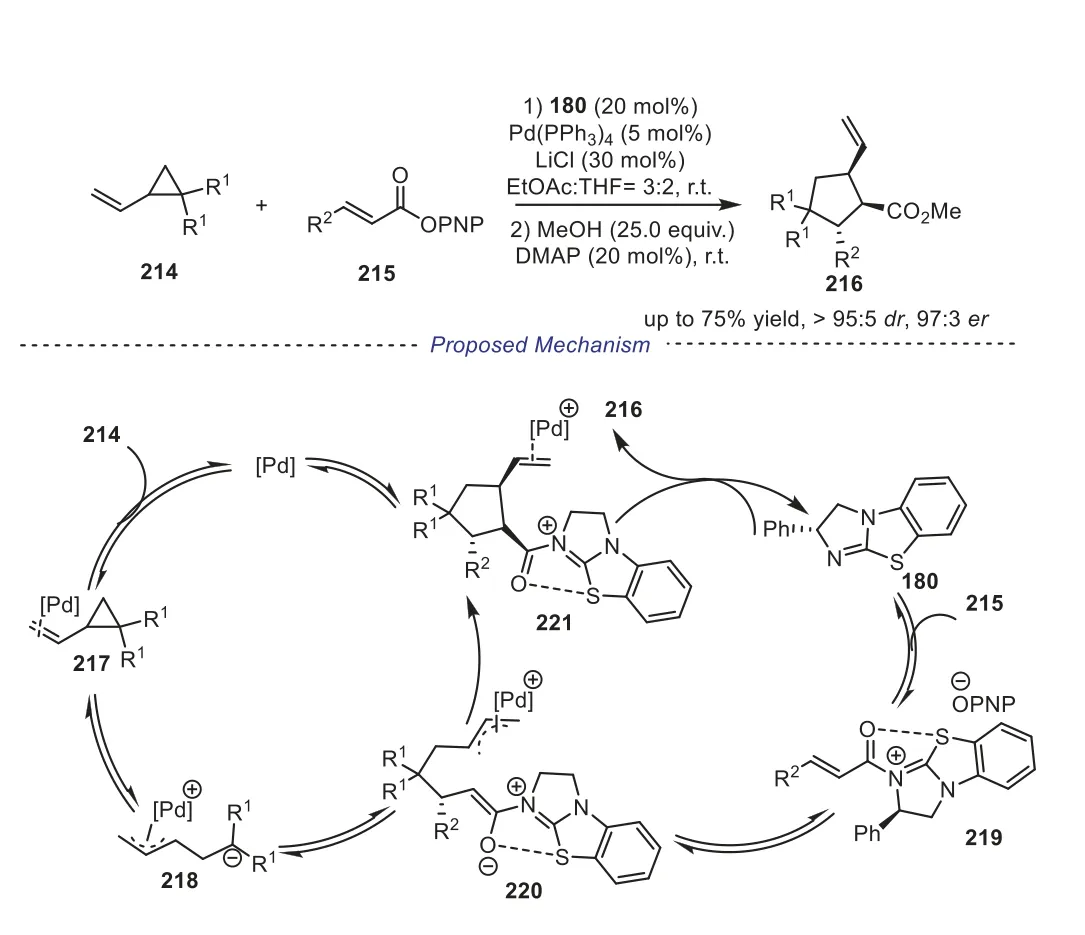
Vinylcyclopropanes have emerged as promising and powerful substrates for palladium-catalyzed [3+2] cycloadditions.In 2022,Smith and coworkers reported an isothiourea/palladium cooperative catalyzed enantioselective [2+3] cycloaddition of vinylcyclopropanes andα,β-unsaturated esters [82].After extensive condition screening, it was found that the optimal conditions required achiral Pd(PPh3)4/chiral isothiourea dual catalytic system in EtOAc:THF (3:2) at room temperature under argon atmosphere, the functionalised cyclopentanes216were generated in good yields and excellent diastereo- and enantioselectivity.It was worth noting that the salt additive (LiCl) played a significant role in obtaining high levels of diastereo- and enantioselectivity because of the addition of Cl-ions generally increases the rate ofπ-σ-πisomerization within Pdπ-allyl intermediates [83–85].The authors proposed a reactive mechanism showed in Scheme 37.Initially,π-allyl-palladium intermediate218was obtained by treatment of vinylcyclopropane214with Pd(PPh3)4.Concurrently, isothiourea180underwent reversibleN-acylation with PNP ester215,givingα,β-unsaturated C1 ammonium intermediate219.Subsequently, continuous Michael addition (220), intramolecular ring closure generated cyclopentane221.Finally, decomplexation of the palladium catalyst and irreversible turnover of the isothiourea catalyst bypara-nitrophenoxide furnished the desired products216(Scheme 37).

Scheme 36.An efficient tertiary amine/iridium complexes cooperatively catalytic strategy for construction of valuable 4-azepane or 4-piperidine motifs.
Scheme 37.Isothiourea/palladium cooperative catalyzed enantioselective [2+3] cycloaddition of vinylcyclopropanes and α,β-unsaturated esters.

Scheme 38.Tertiary amine/Cp*Rh cooperative catalytic C–H bond functionalization.
C–H bond functionalizations have emerged as powerful methods for the construction of valuable organic molecules.More recently, Matsunaga and coworkers disclosed a cooperative catalytic system composed of an achiral Cp*Rh catalyst and a chiral tertiary amine catalyst, for an enantioselective C–H functionalization reaction [86].The experiment result demonstrated that the cooperative catalytic systems were highly tolerant of functional groups to give a wide range of target products225in high enantioselectivities.Notably, the authors found that the slow addition of223might suppress not only its decomposition but also a possible racemic background reaction between a metallacycle intermediate and223,thus increasing the yield and resulting in a significant improvement of the enantioselectivity.In addition, further study suggested that the isomerization of (Z)-223to (E)-223was negligible, which showed that the isomerization of 223 occurred under the reaction conditions and/or that the insertion of the double bond was reversible and the insertion step was less likely to be enantio–determining step (Scheme 38).
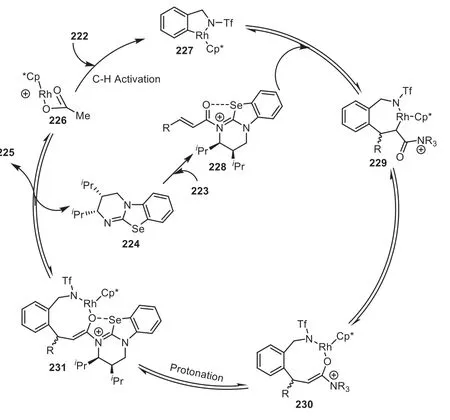
A plausible mechanism for this cooperative catalytic process was outlined in Scheme 39.Initially, a cationic Cp*Rh(III) species226was obtained from [Cp*Rh(CH3CN)3](SbF6)2and KOAc, which reacted with222to form a metallacycle intermediate227.Meanwhile, a C1 ammonium intermediate228was generated by the nucleophilic attack of tertiary amine toα,β-unsaturated precursor223.The authors thought that the conformation of228could be fixed by a noncovalent interaction between the acyl oxygen atom and the selenium atom from the catalyst.Next, the acyl ammonium228coordinated with metallacycle227, followed by the insertion of the C–C double bond to afford C-enolate229, which subsequent furnished a O-enolate230by an isomerization.Finally,protonation of230to give231and subsequent cyclization between the directing group and the acyl ammonium moiety furnished the product225(Scheme 39).
4.2. Tertiary amine / metal relay catalytic annulations
Scheme 39.A plausible mechanism for tertiary amine/Cp*Rh cooperative catalytic C–H bond functionalizations.

Scheme 40.Isothiourea/palladium relay catalytic asymmetric annulation reaction of bromides, CO and ketimine.
Tertiary amine/metal relay catalysis has found widespread applications in organic synthesis.For this type of chemistry, the C1 ammonium enolate generated from the nucleophilic addition of a tertiary amine to a preformed orin situgenerated ketene is pivotal.Thus developing a general route to the catalytic generation of chiral C1-ammonium enolates from simple starting materials has become an active research field.In 2019, Gong and coworkers applied palladium-catalyzed carbonylation reaction to isothiourea/metal reply catalysis [87].Using inexpensive CO as the C1 source, the authors envisioned that a ketene intermediate first generated by palladium-catalyzed carbonylation reaction, which next formed C1-ammonium enolate in the presence of a chiral isothiourea catalyst.Here the authors reported an isothiourea/palladium relay catalytic asymmetric [1+1+4] annulation reaction of bromides232, CO and ketimine233, delivering the desired products235in good yields with high enantioselectivities.This strategy owned well substrate tolerance, andN-tosylimine236could be applied as reactive substrate to give the correspondingβ-lactams237with high enantio– and diastereoselectivities and good yields.In addition, an antiproliferative agent239could be obtained by twostep classical transformations (Scheme 40).
Scheme 41.A plausible mechanism for isothiourea/palladium relay catalytic asymmetric annulation reaction.
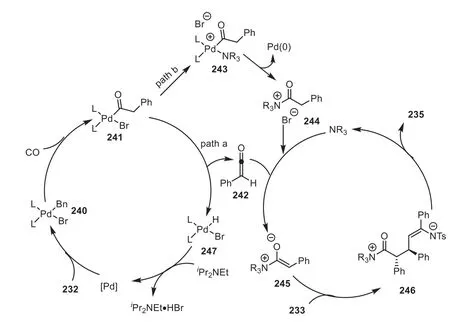
The authors proposed a plausible mechanism, as showed in Scheme 41.Initially, the oxidative addition reaction of palladium catalyst to benzyl bromide232generated the intermediate240,which then underwent an insertion reaction with CO to generate acylpalladium241.Next, the authors thought that two possible paths might occur: one is that the acylpalladium intermediate241bearingα,β-hydrogen might be transformed to the ketene242throughβ-hydride elimination, which then reacted with isothiourea catalyst234to form the key C1-ammonium enolate intermediate245; other alternative pathway was to generate245from acylpalladium241, which coordinated with catalyst234to give an intermediate243, and then generating an ammonium salt244by reductive elimination.In the presence of a base, the salt could transform into ammonium enolate245.Finally, successive Michael addition and intramolecular cyclization generated the corresponding dihydropyridone235(Scheme 41).
5.Conclusions
Dual catalysis has proven an efficient strategy to achieve unprecedented transformations and selectivity that are not available using a single catalyst.The combination of diverse catalysts has been applied widely to the synthesis of fine chemicals, pharmaceuticals, and materials.As described in this review, we have summarized the recent advances on nucleophilic Lewis base/metal dual catalyzed cyclization reactions.The combination of metal with a variety of nucleophilic Lewis base catalysts, such as phosphines,N-heterocyclic carbenes (NHC) and tertiary amines, has enabled a large number of unprecedented cycloaddition reactions by employing appropriate reactive precursors.Obviously, these reported methodologies show a bright prospect to further design and discover new reaction modes and realize challenging chemical transformations that had not been possible using either nucleophilic Lewis base or the metal catalyst alone.
Despite these significant achievements made in this area, there are still great challenges that need to be resolved.For example,catalyst compatibility issues still pose constraints on the effective combination of versatile activation modes.In addition, the catalyst loading of Lewis base required is fairly high (usually 10–30 mol%).Therefore, the development of the design of new, highly active Lewis base catalysts and highly efficient synthetic strategies is becoming more attractive.Undoubtedly, the concept of green chemistry and sustainable development will prompt chemists to develop new avenues to solve these problems, such as performing reactions in aqueous media, lowering the loading of catalysts and developing universal dual catalytic module.As this perspective has illustrated,we hope that, in due time, a general system involving dual catalysis will appear to advance the development of practical organic synthesis in the future.
Declaration of competing interest
We declare that we have no financial and personal relationships with other people or organizations that can inappropriately influence our work, there is no professional or other personal interest of any nature or kind in any product, service and/or company that could be construed as influencing the position presented in, or the review of, the manuscript entitled “Recent Advances in Annulations Enabled by Nucleophilic Lewis Base/Metal Dual Catalysis”.
Acknowledgments
We are grateful to the National Natural Science Foundation of China (No.21702189), the Key Scientific and Technological Project of Henan Province (No.202102310004), Science and Technology Research and Development Plan Joint Fund (cultivation of superior disciplines) Project (No.222301420042) and Zhengzhou University(No.JC21253007) of China for financial support of this research.
 Chinese Chemical Letters2023年10期
Chinese Chemical Letters2023年10期
- Chinese Chemical Letters的其它文章
- Tribute text in memoriam of James N.Seiber (1940–2023)
- Recent advances in MXenes-based glucose biosensors
- Oxidative cyclopalladation triggers the hydroalkylation of alkynes✩
- An integrated supramolecular fungicide nanoplatform based on pH-sensitive metal–organic frameworks
- Probing the effect of nitrate anion in CAN: An additional opportunity to reduce the catalyst loading for aerobic oxidations✩
- Nickel-catalyzed reductive coupling reaction of monofluoroalkyl triflates with alkyl carboxylic acids toward the synthesis of α-alkyl-α-fluoro-alkylketones✩