
Engineering CRISPR/Cas-based nanosystems for therapeutics, diagnosis and bioimaging
2023-11-18 09:27NchunSongShuiLiZhoyueLvXiohuiDingFengLiDyongYng
Chinese Chemical Letters 2023年10期
Nchun Song, Shui Li, Zhoyue Lv, Xiohui Ding, Feng Li,*, Dyong Yng,b,*
a Frontiers Science Center for Synthetic Biology, Key Laboratory of Systems Bioengineering (MOE), Institute of Biomolecular and Biomedical Engineering,School of Chemical Engineering and Technology, Tianjin University, Tianjin 300350, China
b Zhejiang Institute of Tianjin University, Ningbo 315201, China
Keywords:CRISPR/Cas DNA nanotechnology DNA nanomaterials Cancer therapy Diagnosis Bioimaging
ABSTRACT CRISPR/Cas system has been utilized to rationally manipulate intracellular genes, and it has been engineered as versatile and efficient gene editing tools with precise site-specificity and excellent targeting ability for therapeutics, diagnostics, and bioimaging.Here, the evolution and application of CRISPR/Cas systems were sketched chronologically.Landmark works were exemplified to illustrate the design principles of CRISPR/Cas systems.Furthermore, the delivery vectors of CRISPR/Cas system especially DNA nanomaterials-based vectors were categorized and illuminated.DNA nanomaterials are suitable for CRISPR/Cas system delivery via base pairing due to its sequence programmability and biocompatibility.Then the applications of CRISPR/Cas in diagnosis and genomic imaging were highlighted.At the end of the review, the challenges and opportunities of CRISPR/Cas systems were deeply discussed.We envision that the grant advances on CRISPR/Cas systems will promote the development of interdisciplinary fields in chemistry, biology and medicine.
1.Introduction
Since the double helix structure of DNA has been discovered,scientists devoted their many efforts to manipulate gene, in order to explore the secret of life and understand the pathogenic mechanism of diseases [1,2].Gene editing technology usually uses nucleases to induce DNA strand breaks and results in indel mutation in the genomein vivoand several gene editing tools have been developed [3–6].Compared to the first-generation gene editing tools zinc finger nucleases (ZFNs) and the second-generation gene editing tools transcription activator-like effector nucleases(TALENs), the third-generation gene editing tools clustered regularly interspaced short palindromic repeat and associated protein(CRISPR/Cas) system has become more accurate and efficient, and has been wildly used to manipulate the genomes in cells and organisms [7–9].
According to statistics, there are at least 2 classes, more than 30 types Cas protein having been discovered [10–16].And sp-Cas9 belonged to class 2 CRISPR systems comes fromStreptococcus pyogeneswas the most used [8,17,18].The classical Cas9 (Sp-Cas9) mainly included three functional elements: CRISPR RNA (cr-RNA), transactivating CRISPR RNA (tracrRNA) and Cas9 protein.cr-RNA was complementary to the target DNA sequence, and tracr-RNA was partly complementary to crRNA to promote Cas9 nuclease bound to target DNA [19].Particularly, crRNA and tracrRNA were usually combined into a single guide RNA (sgRNA) in laboratory applications for ease of use [17].And Cas9 protein consists of two active endonuclease domain, HNH and RuvC domain.HNH domain induced target strand cleavage, and RuvC domain induces another strand cleavage, generating blunt ends [20,21].
Since being evidenced the capability of targeting and cleavage genome DNAin vitroin 2012 and in eukaryotic cell in 2013,CRISPR/Cas9 has been emerged as a rising star due to the flexibility and versatility properties [8,17,22].To help the readers to clarify the area more clearly, we outlined the development and main applications of CRISPR/Cas in Fig.1 in chronologically [8,10–15,23–38].According to statistics, more than 35 projects about CRISPR technology have come into clinical trials, and some of those were listed in Fig.1 (The date was from www.clinicaltrials.gov by 2022.11.1.).It was predicted that over the next 5–10 years, the therapeutic genome editing will be applied in clinicals [1].However,the off-target effect and efficient delivery of CRISPR/Casin vivoare still concerned now.

Fig.1.The development roadmap of CRISPR/Cas system and its applications in clinical trials and diagnosis.The date of clinical trials was from www.clinicaltrials.gov by 2022.11.1.Development timeline: report of CRISPR clustered repeats [23]; CRISPR widespread in prokaryotes;[24]; ‘CRISPR’coined [25]; Spacer comes from viruses or plasmids[26]; CRISPR mediates adaptive immunity [27]; crRNA guide antiviral defence [28]; Type III-B Cmr CRISPR complexes cleave RNA [29]; tracrRNA directs the maturation of crRNAs [30]; CRISPR/Cas9 cuts DNA in vitro [17]; Genome engineering in eukaryotic cells [8]; Cpf1 (Cas12a) discovery [10]; C2c2 (Cas13a) discovery [11]; Cytidine base editors[13]; Cas X and Y discovery [12]; Adenine base editors [14]; REPAIR (RNA Editing) [31]; Cas14 discovery [15]; xCas9 variant [32]; RESCUE (RNA Editing) [33]; Prime editing[34]; SpG and SpRY variant [35]; very fast CRISPR [36]; Sb-gRAMP [37]; Cas7-11 [38].Diagnosis: CASFISH [40]; NASBACC [41]; SHERLOCK [42]; CUT-PCR [43]; CRISPR-mediated DNA-FISH method [44]; PC reporter system [45]; SHERLOCK v2 [46]; DETECTR [47]; Cas14-DETECTR [15]; HOLMES [48]; CRISDA [49]; CasPLA [50]; HUDSON-SHERLOCK [51];RCH [52]; CAS-EXPAR [53]; MICR [54]; CARVER [55]; CaT-Smelor [56]; CDetection [57]; FLASH-NGS [58]; CRISPR–Chip [59]; RACE [60]; M-CDC [61]; APC-Cas [62]; CUT-LAMP[63]; CASLFA [64]; CARMEN-Cas13 [65]; TL-LFA [66]; FELUDA [67]; opvCRISPR [68].
Efficient delivery of CRISPR/Cas into target cells is vital important for the practical applications, and the advantages and disadvantages of different delivery methods and vectors are compared in many other reviews [6,19,39].We especially emphasized the CRISPR/Cas delivery by DNA nanomaterials due to the sequence programmability, precise base-pairing and biocompatibility of DNA molecules.Further, the non-therapeutic gene editing applications such as nucleic acid and non-nucleic acid detection, and genome imaging were also illustrated simply [15,40–68].Finally, we envision the next generation applications and challenges of CRISPR/Cas in biomedicine, materials science, biotechnology and nanotechnology.
2.CRISPR/Cas system for therapeutics
CRISPR/Cas9 system based therapeutic gene editing has been explored in the clinical trials [9,39,69,70].The naked CRISPR/Cas9 was easily inactivated and degraded in the blood circulation, and furthermore, the difficulties in tissue targeting and cellular uptake made the development of nanocarrier urgently needed for the efficient loading and targeted delivery of CRISPR/Cas9 [19].

Fig.2.The CRISPR/Cas9 system could be delivered as plasmid or liner DNA encoding Cas9 protein and sgRNA, Cas9 mRNA and sgRNA, or Cas9-sgRNA RNP.The CRISPR/Cas9 system could be delivered via three approaches: viral vectors, nano materials, and physical approaches.When delivered as DNA, the system needed transcription in the cell nucleus.The mRNA released in cell nucleus could be translated into Cas9 protein, which combines sgRNA as RNP.The RNP could achieve the function of the therapeutic gene editing in the cell nucleus.
As shown in Fig.2, CRISPR/Cas9 can be delivered in three different forms, including plasmid DNA or liner DNA that encoded Cas9 protein and sgRNA, Cas9 mRNA and sgRNA, or Cas9/sgRNA ribonucleoprotein (RNP).Plasmid DNA is relatively stable and easy to prepare, while plasmid DNA needs enter the nucleus for further transcription, and then translated into Cas9 protein in the cytoplasm,which needs a long time period.Besides, delivery of plasmid DNA has the possibility of integration into the host genome, which may lead to severe damage to organism [71].Compared with DNA,mRNA translation occurred in the cytoplasm, thus avoiding the entrance of nuclear.And due to the mRNA translation was transition,the risk of integration into genome could be largely decreased.Cas9 mRNA contains about 4500 nucleotides, which makes an easy degradation, so the effectiveness of Cas9 mRNA delivery was limited [72].Up to now, the safest and most efficient delivery form was Cas9/sgRNA RNP.Compared with the first two forms,Cas9/sgRNA RNP could enter the nuclei directly without transcription and translation process, which completely circumvents the disadvantages of DNA and mRNA.Furthermore, after cutting the target gene, Cas9/sgRNA RNP could be degraded, which endowed Cas9/sgRNA RNP low immunogenicity, low off-target effect, and high gene editing effect.However, Cas9 proteins could be degraded in the plasm and the persistent expression of Cas9/sgRNA RNP might lead to increased off-target effect.Thus, researches on delivery of RNP for therapy application are emerging.
Physical approaches, viral vectors and non-viral vectors are the most used strategies to deliver CRISPR/Cas into target cells.Physical approaches for delivery of CRISPR/Cas9 system mainly include electroporation, microinjection and hydrodynamic injection.Although these methods are convenient to operate in cell level and widely applied to different cell types, it still has great challengein vivo.Besides, these methods usually only handle a small number of cells per experiment.More importantly, physical approaches may cause severe damage to the cell membrane [73,74].Due to high transfection efficiency and stable transgenic expression, viral vectors have been widely explored to deliver CRISPR-Cas9 systems bothin vivoandin vitro.The commonly used viral vectors included adenoviral vectors (AV), adeno-associated viruses (AAV), lentiviral vectors (LV) and bacteriophages.Although the viral vector could effectively deliver CRISPR/Cas9 system through cell transduction,their clinical applications were hindered by packing size, persistent expression, safety issues and targeting capability.Furthermore, viral vectors often elicited an immune response and caused damage to normal tissues and organs, which even threated life due to the failed targeting [6,75].
Non-viral vectors such as lipid nanoparticles, polymer nanoparticles, hydrogel nanoparticles, inorganic nanomaterials, gold nanoparticles, carbon nanomaterials and cell penetrating peptides have been widely developed to deliver CRISPR/Cas9 system due to their ease of synthesis, lower immunogenicity, excellent targeting ability and multi-functionality [6,76–93].Liposomes facilitated CRISPR/Cas system to cross through the cell membraneviaendocytosis or macrocytosis, and protected from enzymatic degradation and immune clearance.By virtue of excellent DNA affinity, stability in blood and membrane fusion ability, liposomes are emerging as a platform for delivery of CRISPR/Cas system.Liu and co-workers developed a bioreducible lipid nanoparticle simultaneously delivered Cas9 mRNA and sgRNA for fast genome editingin vitroandin vivo[94].Although liposomes presented relatively high transfection efficiency to some extent, the delivery efficiency is highly dependent on cell type and often inhibited by serum.Cationic polymers could effectively compress negatively charged nucleic acidsviaelectrostatic interaction to obtain polymer nanoparticles, which could effectively protect sgRNA and Cas protein from degradation.Chen and co-workers reported a biodegradable nanoparticle for CRISPR/Cas9 system delivery.Taking advantage of the heterogeneous charges’ distribution on the surface of Cas9 protein and sgRNA, various of cationic and anionic monomers, such as imidazole monomer, GSH-degradable crosslinker, acrylate-mPEG and acrylate-PEG-ligand, adsorbed onto RNP through electrostatic interactions first.Thenin situfree-radical polymerization was initiated on the interface of the RNP, realizing single RNP encapsulation in one nanocapsule.CRISPR/Cas9 was could be effectively loaded and delivered in murine retinal pigment epithelium (RPE)tissue and skeletal muscle, which demonstrated high gene editing efficacity [90].
DNA possesses excellent sequence programmability and biocompatibility, and could act as building tiles for constructing DNAbased nanomaterials [95–97].In recent years, DNA nanomaterials have been widely used in disease diagnosis, smart devices, drug delivery, cell culture and cancer therapy.We have successfully used DNA nanomaterials for delivery of Dox, siRNA, and CRISPR/Cas9,which showed great advantages in size-controllability and addressability [98–113].The structure of Cas9 protein and sgRNA allowed the hybridization of sgRNA and DNAviabase complementary pairing, which greatly promoted assembly of Cas9/sgRNA in DNA nanomaterials.And more importantly, the prolonged sgRNA at 3′ terminal showed negligible influences at the combination with Cas9 protein, recognition ability of sgRNA, and cleavage activity of Cas9 protein, which provided a promising strategy for the hybridization of DNA nanomaterials.
Gu and co-workers reported a rolling circle amplification (RCA)-based DNA nanocarrier for the efficient delivery of Cas9/sgRNA RNP [114].In the system (Fig.3A), DNA template was designed with palindromic sequences for the self-assembly of nanoparticles,and base pairing sequences for the combination of sgRNA.The base pairing sequences were set as 12 bp for the efficient delivery of Cas9/sgRNA RNP.After loading Cas9/sgRNA RNP, the cationic polymer polyethylenimine (PEI) was coated on the surface of nanoparticles to form DNA NC, which contributed to promoting endosomal escape.
The zeta potential showed the positively charged Cas9(+19.3 mV), negatively charged Cas9/sgRNA RNP (-19.4 mV).After combination with NC-12, the potential became -28.6 mV, and then recovered to a positive value after coating PEI.Transmission electron microscopy (TEM) images showed a uniform size with 60 nm, demonstrating the successful synthesis of DNC NC.
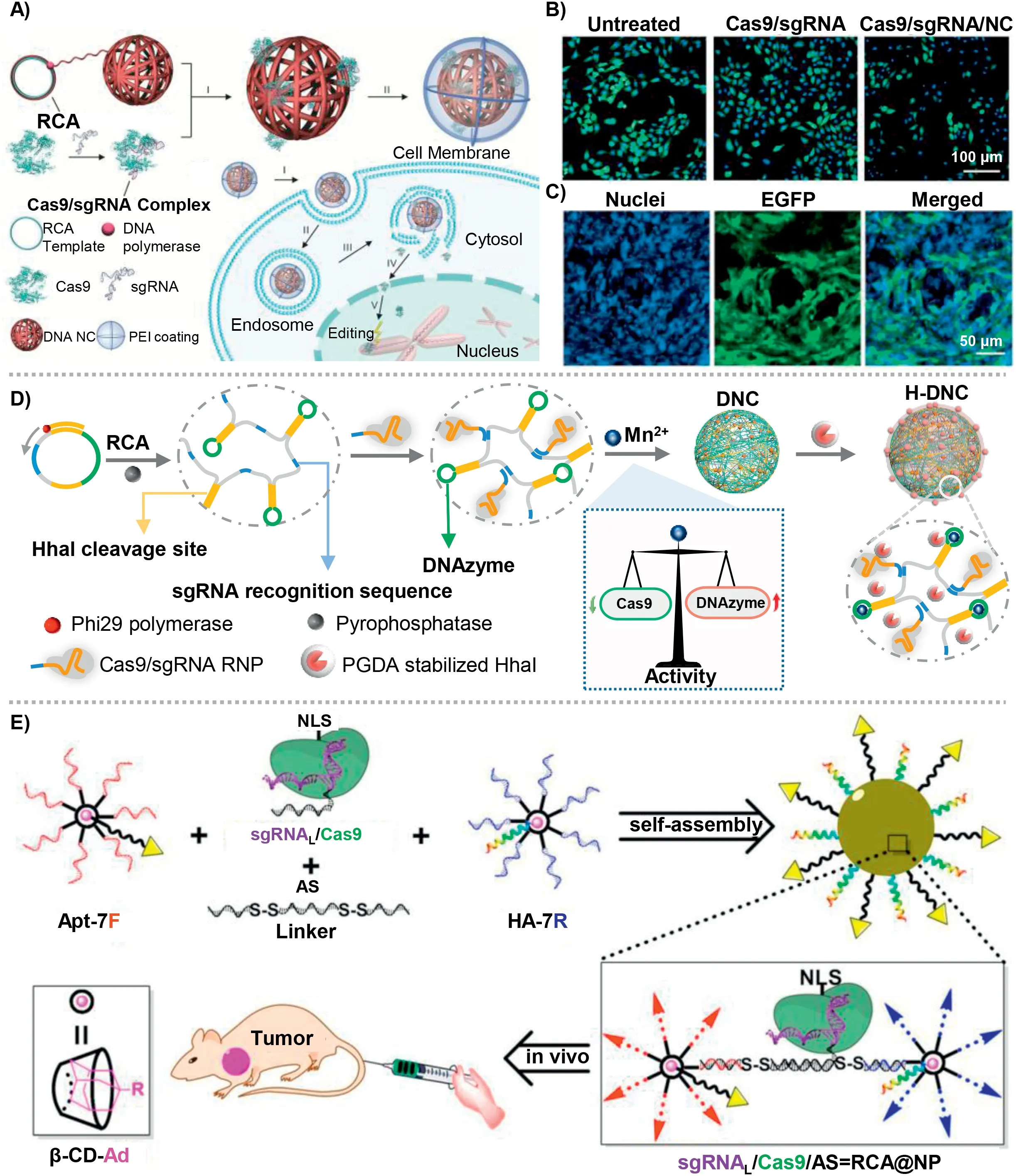
Fig.3.(A) The design of DNC NC via rolling circle amplification (RCA) for the delivery of Cas9/sgRNA RNP.(B) Fluorescence microscopy images of U2OS.EGFP cells with different treatments.(C) In vivo gene editing effect of DNC NC into U2OS.EGFP xenograft tumors in nude mice.Copied with permission [110].Copyright 2015, Wiley Publishing Group.(D) The molecular design and preparation of DNA-based nanosystem for the co-delivery of Cas9/sgRNA RNP and DNAzyme.Copied with permission [111].Copyright 2022, Wiley Publishing Group.(E) Branched DNA nanostructure for the co-delivery of Cas9/sgRNA RNP and ASO for synergistic tumor therapy.Copied with permission [112].Copyright 2019, American Chemical Society.
Furthermore, U2OS.EGFP cells were selected for the visual detection of gene editingviaCas9/sgRNA RNP (sgRNA was designed for targeting EGFP gene).Fluorescence microscopy images showed that compared with untreated U2OS.EGFP cells, Cas9/sgRNA/NC-12/PEI showed the most significant reduction of the fluorescence intensity in approximately 36%, and only Cas9/sgRNA/PEI complex showed only 5% fluorescence reduction (Fig.3B).Thein vivoEGFP disruption effect was performed using U2OS.EGFP tumor-bearing mice.The Cas9/sgRNA/NC-12/PEI showed approximately 25% of the U2OS.EGFP cells showed down-regulated fluorescence intensity,demonstrating the effective gene editing of DNA NC (Fig.3C).On the basis of this work, Gu and co-workers reported another DNA nanomaterials for the delivery of Cas12a/sgRNA RNP for cholesterol regulation in hepatocytes.DNA nanoclew (NC) was producedviaRCA process, for the effective loading of Cas12a/sgRNA RNPviacomplementary base pairing.A layer-by-layer formulation was selected to achieve gene editing efficiency to reduce serum level of cholesterol.The cationic polymer PEI was firstly coated on the surface of NC, turning the negative charge into positive charge, promoting the endosome escape through ‘proton-sponge’effect.Then an anionic polymer layer was coated on PEI, having a charge reversal ability once exposed to acidic environment (anionic to cationic).To enhance the targeted uptake of hepatocytes, galactose was introduced as a hepatocyte-targeted ligand.Using Pcsk9 as a model target, DNA nanocarrier showed efficient gene disruption bothin vitroandin vivo, demonstrating a therapeutic treatment by lowering cholesterol levels.
Recently, we reported a proton-activatable DNA-based nanosystem that enabled co-delivery of Cas9/sgRNA and DNAzyme for the combined gene therapy (Fig.3D) [103].We synthesized an ultralong single-stranded DNAviarolling circle amplification (RCA),including three repetitive functional sequences: recognition sequences of sgRNA, DNAzyme to cleave target mRNA, and HhaI enzyme cleavage sites.We precisely regulated the concentration of Mn2+to balance the catalytic activity of Cas9 and DNAzyme.The concentration of Mn2+was set as 1 mmol/L, which could catalyze the activity of DNAzyme and have negligible influence on the activity of Cas9 protein.The acid degradable polymer (poly-glycerol dimethacrylate)-coated HhaI enzyme was assembled on the surface of nanoparticles, which could response to the acidic environment in lysosomes to recover cleavage activity of HhaI, cutting the target sites in DNA chain to release Cas9/sgRNA RNP and DNAzyme for the subsequent gene editing and gene silencing.Both gene editing by Cas9/sgRNA RNP and gene silencing by DNAzyme showed the synergetic high therapeutic efficacyin vitroandin vivo.Western blotting (WB) was performed to analyse the expression of PLK1 protein (Cas9/sgRNA RNP targeted) and EGR-1 protein (DNAzyme targeted).The results showed that both H-DSCEand H-DECEgroups had the significant down-regulated expression of PLK1 protein,demonstrating the efficient gene editing effect.And both H-DECSand H-DECEgroups showed the significant down-regulated expression of EGR-1 protein, demonstrating the concentration of Mn2+was high enough for the catalysis of DNAzyme.T7EI assay was detected to further access the cleavage effect of target gene.HDECSand H-DECEgroups showed the highly genetic cleavageviaCas9/sgRNA RNP, reaching approximately 40%.The cell viability analysis demonstrated that the combined therapy of gene editing and gene silencing had the best therapeutic effect.Subsequently,thein vivoexperiments further confirmed the efficacy of combined therapy.After an 18-day treatment, the relative tumor volume was curved, the excised tumor xenografts were imaged and weighted.As results showed, the most effective tumor suppression was observed in the final group, and the anti-tumor effect reached approximately 85%, indicating the availability of combined gene editing and gene silencingin vivo.
With the development of DNA nanotechnology, DNA can hybridize with other molecules through chemical bonds, which expands the application of DNA and endows DNA with multifunctional characteristics.Zhang and co-workers reported a nucleic acid nanogel for the delivery of CRISPR/Cas9in vitro[87].In the system,DNA-grafted polycaprolactone brush (DNA-g-PCL) was used to load Cas9/sgRNA RNP through complementary base pairing between grafted DNA and sgRNA, and the DNA-g-PCL was then crosslinked by DNA linker to form a nano-hydrogel [87].Cas9/sgRNA RNP was embedded into the nanogel, achieving physiological stability against nuclease digestion and promoting cellular uptake effi-ciency.Inside cells, the DNA nanogel could be degraded by DNase I for the controlled release of Cas9/sgRNA RNP for the subsequent gene editing.The gene editing efficiency was evaluatedviausing transfection of the EGFP-expression HeLa cells.The sgRNA was designed to target EGFP gene.Confocal laser scanning microscopy (CLSM) images showed that compared with PBS group and Cas9/sgRNA RNP, Nanogel-Cas9/sgRNA showed the most significant down-regulation of green fluorescence, owing to the difficultly cellular uptake ability of naked Cas9/sgRNA RNP.Western blotting and quantitative real-time polymerase chain reaction (qRTPCR) assay demonstrated the down-regulated EGFP protein and mRNA expressions, which was consistent with the results of CLSM image.Furthermore, surveyor assay was performed the cleavage effect of targeted gene in nuclei, which showed approximately 18.7%mutation frequency, demonstrating the effectiveness of nanogelbased CRISPR/Cas9 delivery systemin vitro.
Besides, Ding and co-workers constructed a branched DNA nanostructure for the co-delivery of Cas9/sgRNA RNP and antisense oligonucleotide (ASO) (Fig.3E) [115].β-Cyclodextrin was modified with azide for the construction a DNA motif with seven DNA branches.Then, adamantine-conjugated aptamer and influenza hemagglutinin peptide was introduced to endow the DNA nanostructure with targeting ability and endosomal escape ability.Furthermore, a multi-functional DNA linker was rationally designed to construct the finally DNA nanostructure loaded with Cas9/sgRNA.The DNA linker combined ASO sequences and complementary sequence of DNA motif connected by disulfide bond connected.More importantly, the extend sequence of sgRNA was complementary to ASO, which enabled loaded Cas9/sgRNA into the DNA nanostructure.After uptake by targeting cells, responding to intracellular glutathione (GSH) and RNase H, DNA nanostructure disassembled and controlled release of Cas9/sgRNA RNP and ASO for further synergistic tumor therapy.MCF-7-EGFP cells was chosen to visually verify the gene editing efficiency.CLSM images showed that RECAE@NP group (sgRNA was designed to target EGFP gene, ASO was designed to target EGFP mRNA) significantly downregulated the expression of EGFP protein, contrast with RSCAS@NP,RSCAE@NP, and RECAS@NP groups.The results of Western blotting were consistent with CLSM.Furthermore, T7EI assay was detected to verify the gene cleavage efficiency.Both RPCAS@NP and RPCAP@NP showed similar cutting efficiency, demonstrating the efficient gene editing effectin vitroof DNA-based nanoplatform.MCF-7-EGFP tumor xenograft model was established for the visualization of gene editingin vivo.A marked reduction of green fluorescence was observed in RECAE@NP group.MCF-7 tumor-bearing mice was further established to verify the effect of synergistic gene therapy of DNA-based platform.The RPCAP@NP group (synergistic gene therapy of gene editing and gene silencing) showed the best antitumor effect.In addition to organic molecules, inorganic materials can also bind to DNA for the delivery of Cas9/sgRNA RNP.Le and co-workers designed a termed binding-mediated protein corona (BMPC) for the delivery of Cas9/sgRNA RNP.AuNPs and magnetic nanoparticles (MNPs) were mixed to construct scaffold.Thiol-labelled oligonucleotide (SH-DNA) with a binding site was combined to AuNPs with gold-thiol bonds.A ternary complex containing Cas9 protein, sgRNA, and linker DNA can bind to SH-DNAviacomplementary base pairing, which had photo-cleavable (PC)linkers with sgRNA at 5′ terminal.After UV illumination, PC linkers were broken for the controlled release of Cas9/sgRNA RNP.
Despite various carriers and great advance have acquired in recent years, delivery of CRISPR/Cas systemin vivoremains great challenges.Not only should the biocompatibility of carrier be considered, the efficient loading capability and effective release of CRISPR/Cas without affecting its activity is also vital important.Compared with DNA and mRNA, direct delivery of RNP facilitates more efficient gene editing, reducing off-target effects and lowering the risk of integration into the genome, which showed great potential in clinical trials.DNA nanomaterials could be developed as a common platform to deliver gene editing tools, promoting the advances in the clinical trials at genetic level.
3.CRISPR/Cas system for diagnostics
Nucleic acid detection in medical diagnostics is crucial for identification and prevention of many diseases, but remains great challenges due to the sensitivity and accuracy [116].qPCR, RNA-seq,high-throughput immunoassays and mass spectrometry (MS) tests are the most used methods in clinical.However, they are often time intensive, costly and dependent on sophisticated equipment and well-skilled technical staff [117].Therefore, it is vital to develop novel nucleic acid detection technologies in resource-limited application scenarios.
As the adaptive immune systems in archaea and bacteria,CRISPR/Cas has capability of precise recognition and quick response to target nucleic acids [118].Therefore, the CRIPSR/Cas has been deeply explored for precise detection of nucleic acid.Apart from Cas9, other Cas protein with collateral cleavage activity, such as Cas12a, Cas13a and Cas14, are more convenient in DNA and RNA detection (Figs.4A and B) [119,120].The Cas effector exhibits a non-specific nucleases activity which is also termed as collateral cleavage.After activation by target sequence, they could promiscuously cleave ssDNA, ssRNA or dsDNA.Combined with other nucleic acid amplification methods, such as strand displacement amplification and recombinase polymerase amplification (RPA), the sensitivity and accuracy of CRISPR involved amplification system were greatly increased.The main CRISPR-based diagnostic system have been summarized in Fig.1.
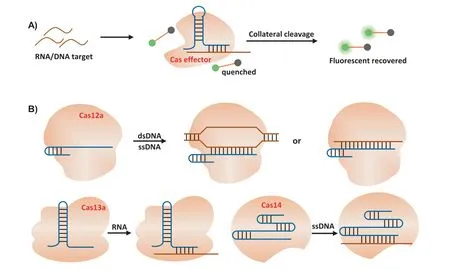
Fig.4.(A) Schematic diagram of detection strategies based on the “collateral” cleavage of the Cas effectors.(B) Cas12a, Cas13a and Cas14a effector were activated by different nucleic acid target.Cas protein is inactive in the absence of the target sequence.Once guide RNA binding its DNA/RNA target, the “collateral” cleavage activity of Cas protein to nontarget ssDNA (Cas12a or Cas14a) or ssRNA (Cas13a) is activated, leading to the digestion of the quenched fluorescent ssDNA/ssRNA reporters and the release of the fluorescent signal that indicates the presence of the target DNA/RNA.dsDNA, double-stranded DNA; ssDNA, single-stranded DNA; ssRNA, single-stranded RNA.
Cas9 could be used for pathogen genotyping and single nucleotide polymorphism (SNP) identification combined with nucleic acid amplification methods.Pardee and co-workers combined nucleic acid sequence-based amplification (NASBA) with Cas9 effector for the genotyping of Zika virus RNA [41].Zhou and co-workers developed CRISDA (CRISPR-Cas9-triggered nicking endonucleasemediated Strand Displacement Amplification) for human breast cancer genotyping in attomolar sensitivity and single-nucleotide specificity [49].The Cas9 with a mutation of HNH catalytic residue was employed to recognize the target DNA and induce a pair of nicks in both non-target strands.That made it possible for the primer pairs to hybridize with the exposed non-target strands and start strand displacement amplification.Finally, the amplicons were quantitatively determined by measuring the fluorescence intensity of invasive peptide nucleic acid.
Lateral flow assay is a convenient testing technique, because it can be seen with the naked eye without complex equipment.Wang and colleagues introduced CASLFA (CRISPR/Cas9-mediated lateral flow nucleic acid assay) to identify genome samples [64].The target DNA was firstly amplified using biotin-labelled primers.Then, the biotinylated amplicons were incubated with the designed Cas9/sgRNA and trickled on the sample pad.Under capillary force,the Cas9/sgRNA-biotinylated amplicons moved forward, conjugated with AuNP-DNA probe and were collected by the precoated streptavidin at the test line to generate colored band.The detection sensitivity was as low as 150–200 copies/reaction.
Compared with Cas9, Cas12 effectors possess collateral cleavage activity that led to the development of the DETECTR (DNA Endonuclease Targeted CRISPR Trans Reporter) method.After the process of RPA, CRISPR/Cas12a targeted DNA and trans-cleaved collateral FQ-ssDNA reporter, releasing fluorescent signal readout.This method presented a great ability for accurate and rapid detection of human papillomavirus (HPV) [47].
Inspired by the “collateral effect” of Cas13a upon RNA target recognition, Zhang’s group established a detection platform which was termed as SHERLOCK (Specific High Sensitivity Enzymatic Reporter UnLOCKing), providing rapid DNA or RNA detection with attomolar sensitivity and single-base specificity [121].The CRISPR/Cas13a biosensing system had been successfully applied for detecting specific strains of Zika and Dengue viruses, bacterial, human DNA genotypes, and cancer mutation [42].Intriguingly, SHERLOCK was further improved (SHERLOCKv2) to allow for multichannel detection with higher signal sensitivity and lateralflow readout [46].
Besides the application for nucleic acid detection, Cas12a has been developed as high throughput platforms for detection of small molecules or pathogenic bacteria [62,122].For example, CaTSMelor (CRISPR-Cas12a- and aTF-mediated small molecule detector) had been improved to test uric acid in human serum samples successfully [56].The Cas12-targeted dsDNA contained a motif that was recognized by the cellulose binding domain-allosteric transcription factor (CBD-aTF).When the target small molecules presented and bound with aTF, the conformational change occurred,resulting in the release of dsDNA.Therefore, target small molecules could be quantitatively analysed by measuring the change in the fluorescent signal produced by cleaved FQ-labelled ssDNA probe.Shen and colleagues presented a detection system (called ‘APCCas’) that could selectively and sensitively quantifySalmonella Enteritidiscells without isolation [62].In the system, a specific allosteric probe (AP) was used to recognize target pathogen.When AP specifically recognized and bound with the target pathogen, the conformation of AP changed.The activated probe acted as a template to yield a dsDNA with the help of primer and DNA polymerase.At the same time, the pathogen was displaced from the probe to join another cycle for secondary amplification.Subsequently, numerous target ssRNAs were transcribed from the dsDNA by T7 RNA polymerase.Finally, the ssRNAs activated the collateral cleavage ability of Cas13a to cleave multiple RNA reporter probe,generating fluorescence signals.
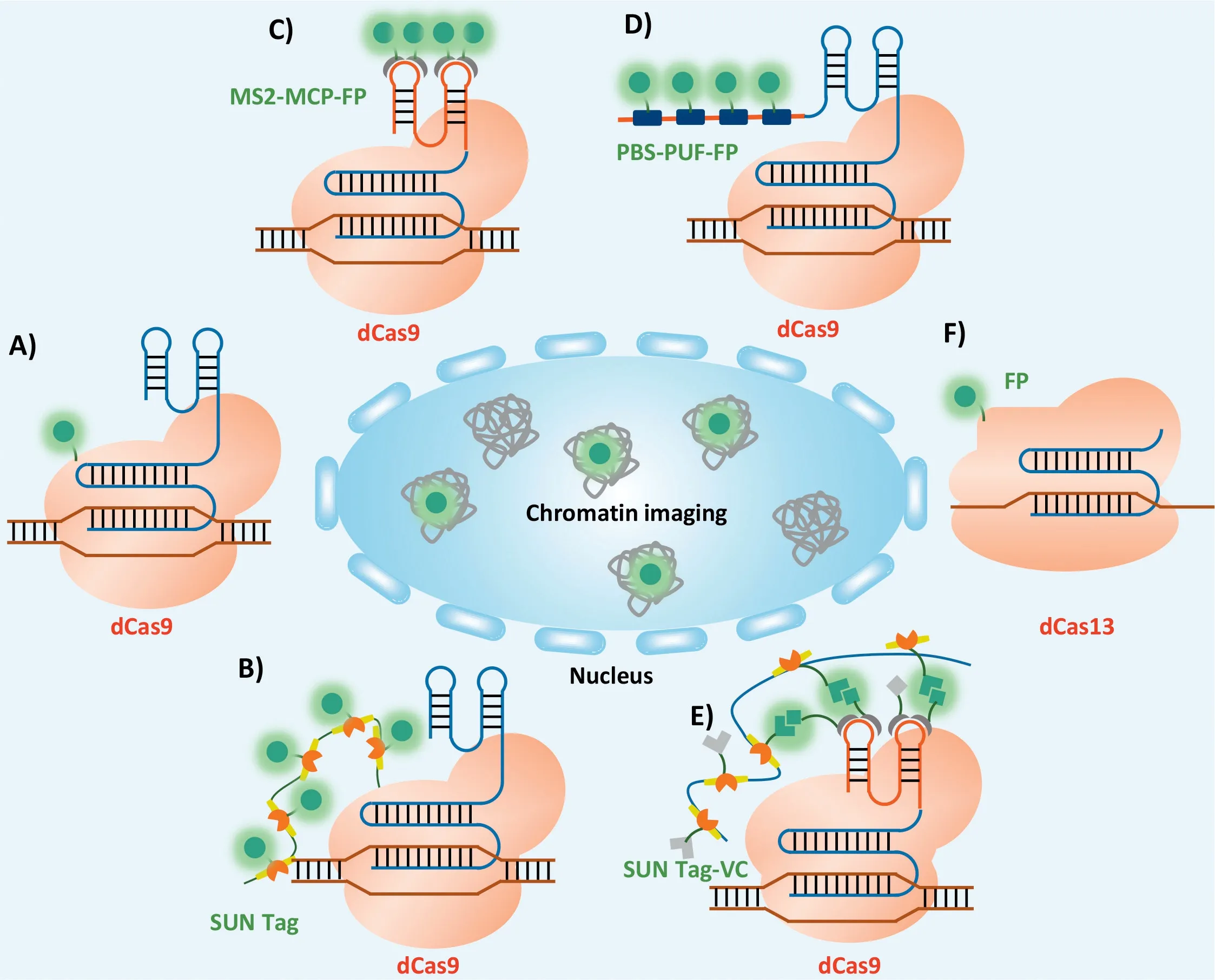
Fig.5.CRISPR-based imaging system.(A) Schematic description of single FP labeling dCas9 imaging system.(B) Schematic description of the multiple FPs labeling dCas9 imaging system.Schematic description of the sgRNA labelled with FPs through RNA aptamer (C) or unique target sequence (PBS) (D).(E) Schematic description of the dCas9 labelled with multiple Venus C-terminal (VC) fragments and sgRNA labelled with multiple Venus N-terminal (VN) fragments.(F) Schematic description of single FP labeling dCas13 imaging system.
The reported CRISPR/Cas biosensing systems have advantages of simplicity to operate, high resolution, high sensitivity, and low cost at a single-base resolution and sub-attomolar sensitivity.CRISPRbased diagnostic devices could be easily modified to detect any target nucleic acid sequence by simply changing the sgRNA sequence,thereby having the potential that could be readily adapted to detect infection from emergent viruses.The CRISPR/Cas systems have great potentials as an important supplement to qPCR and other gold detection standards in clinical applications.
4.CRISPR/Cas system for bioimaging
Genome organization, localization and dynamics are closely relevant to many cellular processes including DNA replication, DNA damage repair and gene expression [123].For instance, the intracellular spatiotemporal localization of specific genomic loci, noncoding RNAs and mRNAs, has been proven to be pivotal to their functions [124].However, the convenient and efficient living cell imaging of different genome, chromosomes and their spatiotemporal relationships is still an enormous challenge.The widely used genome loci imaging method is fluorescentin situhybridization(FISH), wherein an exogenous fluorescent oligonucleotide probe is used to hybridize with specific genome locus [125,126].FISH helps us more deeply investigate the genome structure.However,the materials used in FISH need to be fixed and the hybridization condition easily disrupted the integrity of the materials, which greatly impeded its application in living cells.Due to the excellent DNA/RNA targeting ability and nuclear localization sequence (NLS)-guide nuclear membrane penetration ability, CRISPR/Cas9 system has been explored to label specific genome loci for various applications, including genome loci imaging in living cells [127].Catalytically dead version of the Cas9 (dCas9) loses the ability of gene cleavage, but retain the DNA sequence target and binding ability,so the dCas9 is suitable for non-gene editing applications such as gene imaging [128–130].
In 2013, Chen and co-workers firstly reported using an EGFPtagged dCas9 and sgRNA to image genome locus in living human cells.The CRISPR system enabled imaging the repetitive elements in telomeres and protein coding gene like Mucin.Furthermore, the authors used an array of sgRNAs to image non-repetitive regions in human genome (Fig.5A).Using the system, the telomere dynamics during elongation, the localization of the gene loci, and their dynamic behaviors during mitosis could be visualized [131].However, for non-repetitive sequences, dCas9 has the relatively low signal-to-noise ratio in imaging distribution of specific sites in the genome [127].Some researchers were devoted to tagging dCas9 with more fluorescent proteins (FPs) to decrease the background signal.SunTag adds multiple tiny peptide epitopes to the target dCas9, allowing to add multiple tags to the protein [132,133].These epitopes can recruit fluorescently labelled single chain antibody scFv.For example, applying to SunTag, a poly-general control noninducible 4 (GCN4) peptide scaffold was developed to recruit 24 copies of GFPviathe interactions between GCN4 and the singlechain variable fragment (scFv) (Fig.5B) [132].Using dCas9-SunTag,non-repetitive regions of the MUC4 gene was tracked with only 20 different sgRNAs.
In addition to dCas9-FPs fusion proteins, many researchers have demonstrated the imaging feasibility of modifying sgRNA to recruit more sequence-specific RNA binding proteins labelled by FPs.The RNA aptamer MS2, derived from bacteriophage, would specifically bind to MS2-coat protein (MCP)viaits folded structure.When the MS2 was modified into sgRNA and dCas9 was labelled with MCPFP, the system could be used to image the specific genome locus in living cells (Fig.5C) [134].Mazhar Qin and corkers developed a method that could real-time track genes in living cells, and thus the localization and movement path of genes could be imaged from a 3D perspective.They connected 16 repeated MS2 RNA fragments with the dCas9 at the tail of the single-guide RNA (sgRNA)to avoid destroying the target gene.This method overcame the long-standing limitations of genome imaging and directly observed live cells at the single-cell level, thus helping scientists to observe how specific genes work and interact with a new perspective [135].Another method that named Casilio, was also developed for chromosome imaging.In this system, the dCas9-FPs fusion protein was combined with the Pumilio/Fem3 mRNA-binding factor (PUF), that was, the scaffold structure of sgRNA was modified to the binding region of PUF protein (PBS) and recruiting more FP-PUF fusion proteins (Fig.5D) [136].Despite these advances, the intrinsic nonspecific labelling issue of sgRNA labelling strategy and the high background signal of unbound dCas9-SunTag are still big challenges.Accordingly, a strategy bimolecular fluorescence complementation which combined the advantages of both dCas9 and sgRNA-labelling strategies was proposed.In details, functional Venus molecule was split into C-terminal (VC) and N-terminal (VN) firstly, then scFv-VC was transfected with SunTag-dCas9 and MCP-VN was transfected with MS2 modified sgRNA, the functional Venus molecules could only be fused within dCas9/sgRNA complexes to emit a signal upon excitation, showing a high specific labelling foci of dCas9 imaging system (Fig.5E) [137].Compared to single-labelling, the BiFC demonstrated higher signal-to-noise ratios and increased the effectiveness and specificity of dCas9-based genome imaging.
In addition, Abudayyeh and co-workers confirmed that CRISPR/Cas13 could target RNA in mammalian cells [138].CRISPR/Cas13 system was the RNA targeting endonuclease system mediated by sgRNA.The programmable RNA targeting capability of the Cas13a system could be used to inhibit cellular RNA targets, bind and enrich RNA, and image RNA within cells through sequence-specific binding.For example, Yang and co-workers reported that dCas13 system could effectively label non-coding RNA and mRNA in living cells (Fig.5F) [139].They screened out dPspCas13b and dPguCas13b proteins with better RNA labelling capacity after studying the enzymatic mutations of various CRISPR/Cas13 proteins.Then, they used the optimized CRISPR/Cas system achieved in real-time imaging.Combining the CRISPR-dCas13 system and the CRISPR/dCas9 system, researchers achieved simultaneous labelling of RNA and gene loci transcribed by genes, providing a simple and convenient way to study the relationship between RNA transcription and RNA and gene loci.A robust CRISPR-based imaging system enabled reveal new insights into how chromatin structure and dynamics affect cell function in normal and disease states.
Despite the exciting achievements made in CRISPR-based imaging, it is still unclear whether positioning large exogenous RNP in transcripts interferes with cellular processes.Moreover, more sensitive and specific CRISPR/Cas system is needed for genome imaging for future study.
5.Conclusions
CRISPR/Cas system has revolutionized the field of gene editing,and shows great potentials in therapy, diagnosis, bioimaging, and other fields.As CRISPR/Cas-based technology has entered the stage of the clinical trial, they have great potential for enhancing cell therapies and reversing genetic diseases.In the process of clinical treatment, it is necessary to assess its potential toxicity, immunogenicity, off target effect and the possibility of host genome integration.Developing biocompatible carriers with improving detection sensitivity and circumventing potential risks are of great significance for the clinical applications of CRISPR/Cas system.
There are four barriers for CRISPR/Cas9 system entering the target cells and leading to desired gene editing.The first barrier is various nucleases and proteases in plasma.Packaging the delivery vectors and chemically modifying the backbones of sgRNA and DNA templates has been reported to be effective strategies to improve the stability in the plasma [140,141].The second barrier is clearance by the immune system and endothelial system[142,143].Appropriate particle size and surface properties could reduce the clearance, thus prolonging the circulation timein vivo.The third barrier is to enhance the accumulation of CRISPR/Cas9 system in the target tissue.Targeted tissue-specific ligands could be introduced, the size of the nanoparticles should be optimized,and physical or biological methods could be used to increase the permeability of local vessels transiently [144–146].The last barriers is to escape from endosome and enter the cell nucleus.DNA nanomaterials could be programmed to cater for the requirements in different transport stagesin vivo, such as high stability, targeting capability, controlled release property inside cells and satisfactory biodegradability, therefore, the DNA nanomaterials are emerging as delivery systems of CRISPR/Cas system for various clinical applications.
Due to the specific nucleic acids targeting ability, CRISPR/Cas could be easily designed to detect or image any target nucleic acids including DNA and RNA.The CRISPR/Cas systems have realized single-base detection and sub-attomolar sensitivity.Quick detection with high sensitivity and accuracy makes CRISPR/Cas systems an important supplement to qPCR in clinical detection of nucleic acid.Moreover, scientists have developed CRISPR/Cas-based smart materials, such as a CRISPR/Cas12a responsive DNA hydrogel that realized cargo release from DNA hydrogel.These innovative works demonstrate that CRISPR/Cas has an immense potential in bioanalysis.Although Cas9 was widely researched, evolutive CRISPR systems are still needed to be discovered and developed,which may provide new vitality for a wider range of applications.We envision that the CRISPR/Cas system will make great contributions in biotechnology, chemical science, clinical medicine and materials science.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported in part by National Natural Science Foundation of China (Nos.22225505, 21621004, 21905196 and 31971305), Tianjin Natural Science Foundation (Basic Research Plan,No.18JCJQJC47600), and National Key R&D Program of China (Nos.2018YFA0902300, 2019YFA0905800).
 Chinese Chemical Letters2023年10期
Chinese Chemical Letters2023年10期
- Chinese Chemical Letters的其它文章
- Tribute text in memoriam of James N.Seiber (1940–2023)
- Recent advances in MXenes-based glucose biosensors
- Oxidative cyclopalladation triggers the hydroalkylation of alkynes✩
- An integrated supramolecular fungicide nanoplatform based on pH-sensitive metal–organic frameworks
- Probing the effect of nitrate anion in CAN: An additional opportunity to reduce the catalyst loading for aerobic oxidations✩
- Nickel-catalyzed reductive coupling reaction of monofluoroalkyl triflates with alkyl carboxylic acids toward the synthesis of α-alkyl-α-fluoro-alkylketones✩