
钙和羟基影响焦炭还原N2O的量子化学研究*
2023-11-22 02:01尹艳山邓文鹏陶建航杨茹帆卿梦霞孙刘涛
煤炭转化 2023年6期
尹艳山 邓文鹏 陶建航 杨茹帆 张 巍 卿梦霞 刘 磊 孙刘涛 刘 亮
(长沙理工大学能源与动力工程学院,410114 长沙)
0 引 言
我国是世界上第一大煤炭消费国,N2O是燃煤流化床锅炉排放的主要污染物之一[1]。燃煤排放的N2O会引起酸雨、光化学烟雾、温室效应等一系列环境问题[2-3]。因此,如何减少N2O的排放是燃煤利用领域一个关键问题。焦炭对N2O起着异相还原作用,焦炭表面的活性位将N2O还原为N2[4-5]。此外,大量研究表明,金属氧化物对NOx有直接催化还原作用[6-8]。CHEN et al[9]发现Ca的加入促进了NOx前驱体形成,并提高了煤焦的表面活性。LIU et al[10]通过第一性原理计算发现,Ca的加入增加了NO在石墨烯表面的吸附能。ZHANG et al[11]模拟研究了Ca修饰后焦炭对NO异相还原的影响,发现Ca的加入缩短了NO异相还原的反应路径。HOU et al[12]测定了四种金属化合物(SiO2,Al2O3,CaO和Fe3O4)直接分解N2O的能力,其中CaO和Fe3O4的催化分解作用相对较强,但O2存在时金属氧化物分解N2O的能力降低。WU et al[13]研究发现CaO表面硫酸化程度增加导致其表面活性降低,其分解N2O的能力也大大降低。尽管如此,关于Ca影响焦炭还原N2O机理的理论研究还较鲜见。
含氧气体对焦炭还原NOx有重要影响。黄增辉等[14]发现O2的加入显著抑制了焦炭对NO的还原,氧浓度对焦炭还原N2O存在影响作用,在1 173 K~1 273 K条件下,氧气浓度越高,焦炭还原N2O的能力越强,当温度高于1 273 K时,高氧浓度反而削弱了焦炭还原N2O的能力[15-16]。CHEN et al[17]探讨了氧浓度对NO异相还原的影响,在分子水平上揭示了氧对NO—C反应的影响机制。ZHAO et al[18]研究了在焦炭氮与NO相互作用过程中N2O的形成机理,结果表明,氧分子吸附到焦炭表面后降低了N2O在焦炭表面的解离能。LI et al[19]研究发现提高氧气浓度有助于降低N2O的排放。JIAO et al[20]发现CO在NO异相还原中的催化作用体现在活性位点的还原速率和数量上。CHEN et al[21]基于Zigzag和Armchair焦炭模型发现CO对N2O的均相还原反应还会受到焦炭的催化作用。GAO et al[22]发现CO的加入显著降低了N2O-C反应后期C(O)解吸阶段的能垒,焦炭边缘结构对N2O还原反应的能垒影响较大。此外,焦炭表面的含氧官能团如羟基、羰基、醌基和羧基等亦对焦炭吸附气体有重要的影响[23-25]。ZHANG et al[26]利用密度泛函理论(DFT)研究发现,羟基和羰基的存在增强了焦炭异相还原NO的能力。尽管如此,关于含氧官能团影响焦炭还原N2O的理论研究还较鲜见。
本实验基于密度泛函理论(DFT),模拟研究了原始Zigzag焦炭模型及钙和羟基修饰后的焦炭模型还原N2O的过程,分析了反应过程中的过渡态和中间体,研究了不同原子键的键长、键级、能垒和释放能量的变化,揭示了钙和羟基以及反应温度对N2O-C反应的影响。
1 模型选择与计算方法
1.1 焦炭模型
由于煤焦的分子结构非常复杂,由大量芳香环组成[27-28],通常采用简化焦炭模型研究焦炭还原NOx的机理和影响因素[29-30]。CHEN et al[31]基于含有6个和6个以上芳香环的焦炭模型,使用从头算法进行分子轨道研究,发现得到的参数(键长、键角、拉曼光谱频率等)与实验数据吻合良好,并发现继续增加焦炭模型的芳香环数量到7个以上并没有收到预期效果。此外,随着焦炭模型中苯环数量增加,能量损失波动较小,热释放对物理模型不敏感。因此,Zigzag焦炭模型被广泛用于煤焦气化反应[32-33]、煤焦吸附气体[34-35]和焦炭还原NOx[36]的机理研究,结果表明,Zigzag焦炭模型的边缘位点非常活跃,焦炭还原NOx反应很容易发生在这些位点上[37-39]。本研究采用Zigzag焦炭模型研究钙和羟基对焦炭吸附及还原N2O的影响。原始焦炭模型及钙和羟基修饰的焦炭模型结构如图1所示,N2O在不同焦炭模型活性位点上吸附的稳定结构如图2所示。
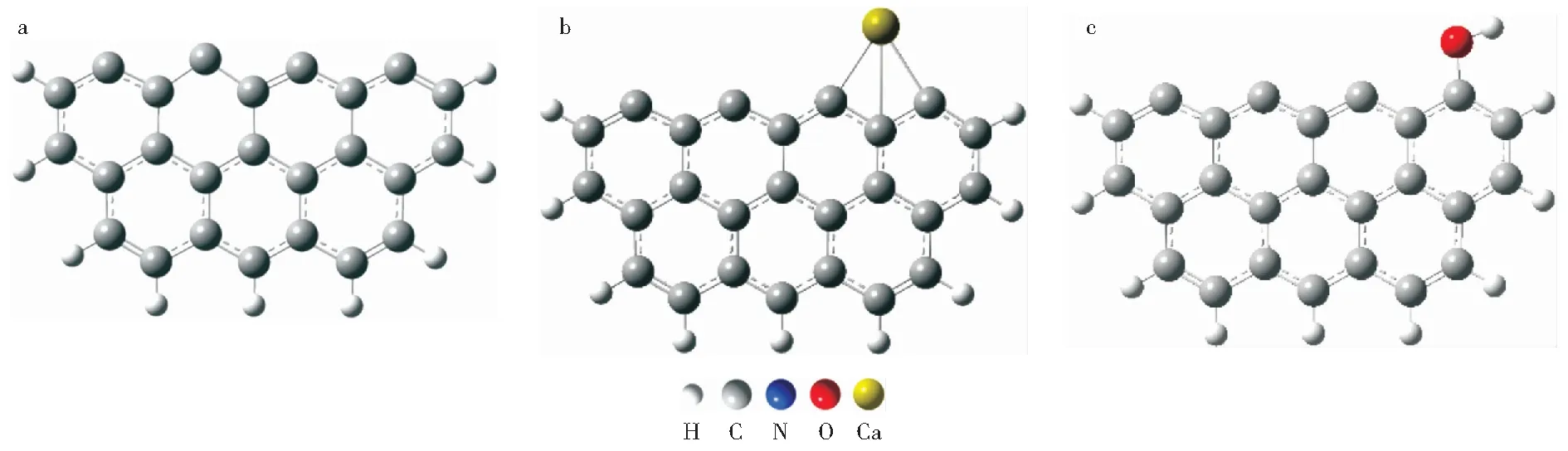
图1 不同焦炭模型的几何构型Fig.1 Geometric structure of different char models a—Pristine Zigzag char model;b—Ca-decorated Zigzag char model;c—OH-decorated Zigzag char model
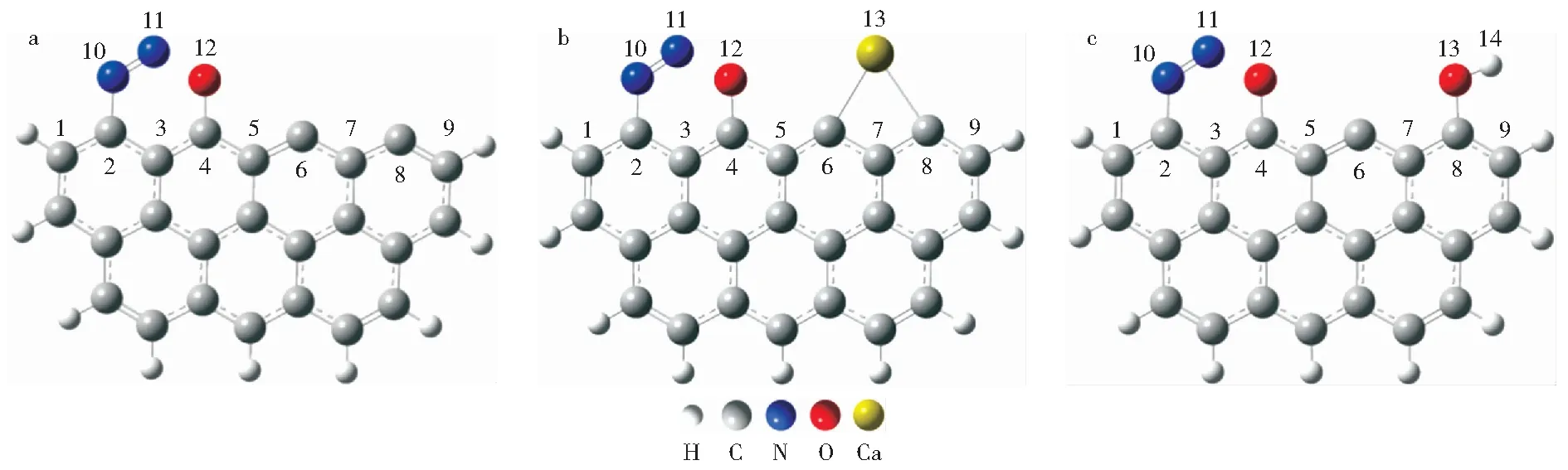
图2 不同焦炭模型吸附N2O的稳定结构Fig.2 Stable structure of N2O chemisorption in different char models a—Pristine Zigzag char model;b—Ca-decorated Zigzag char model;c—OH-decorated Zigzag char model
1.2 计算方法
M06-2X方法已广泛应用于焦炭CO2气化和焦炭还原NO反应的DFT研究,并可获得优化的分子结构[32,40-41]。M06-2X泛函不仅可以最大限度地减少低自旋污染的影响,还可以提高能量势垒和非共价分子间作用力的准确性[42]。在同一理论水平上计算单点能,并进行零点能校正。过渡态有且仅有一个虚频,且虚频对应的原子振动方向与反应趋势一致。因此,本研究在M06-2X/6-311G(d)基组水平上对各势能面的几何结构进行优化和频率分析[43],得到了反应物(R)、生成物(P)、中间体(IM)和过渡态(TS)的构型。为保证过渡态计算的准确性,采用本征反应坐标(IRC)对计算结果进行了校核。所用计算程序为Gaussian 16。
采用过渡态理论(TST)对298.15 K~1 500.00 K温度下N2O还原的动力学参数进行研究,其反应速率常数(kTST)的计算见式(1)[44]。
(1)
式中:kB为玻尔兹曼常数,J/K;T为反应温度,K;h为普朗克常数,J·s;QTS,QA和QB分别为过渡态、反应物A和反应物B的配分函数;E为反应能垒,kJ/mol;R为通用气体常数,8.314 J/(mol·K);Γ为量子隧道修正因子,其经验表达式见式(2)。
(2)
式中:νm为过渡态的虚频率,cm-1;c为光速,m/s。
各基本反应的动力学速率常数k(T)见式(3)。
k(T)=Aexp(-Ea/RT)
(3)
式中:A为指前因子,s-1;Ea为活化能,kJ/mol。
2 结果与讨论
2.1 原始Zigzag焦炭模型还原N2O
原始Zigzag焦炭模型还原N2O过程优化的几何构型和势能面分别如图3和图4所示。由图3可知,此过程共经历5个过渡态(分别为TS0,TS1,TS2,TS3,TS4)和4个中间体(分别为IM1,IM2,IM3,IM4)。焦炭还原N2O可以分为三个阶段:N2O的吸附、N2的析出和C(O)配合物的解吸。首先,N2O分子接近焦炭表面,以N—O侧向的形式吸附到焦炭表面的活性位点,经历过渡态TS0生成中间体IM1。由图4可知,Zigzag焦炭吸附N2O的能垒为83.5 kJ/mol,高于CHEN et al[21]的结果(32.9 kJ/mol),可能的原因为本研究的计算方法(M06-2X泛函)与文献[21]的计算方法(B3LYP泛函)不同。中间体IM1的形成释放出350.6 kJ/mol能量,这在热力学上有利于后续反应。由于N11和O12之间距离增加,形成了更稳定的中间产物IM2,该过程的能垒(3.6 kJ/mol)和能量释放(69.7 kJ/mol)均较低。N11继续远离O12,C2—N10键长增加(0.134 nm→0.139 nm→0.145 nm),N10—N11键长也增加(0.113 nm→0.115 nm→0.117 nm)。同时,C2—N10—N11的键角逐渐减小。经历过渡态TS2形成中间体IM3,需要克服的能垒较低(9.2 kJ/mol)。接着N10—C2键长增加(0.145 nm→0.182 nm→∞,∞表示超出成键范围),N10—N11的键长减小(0.117 nm→0.112 nm→0.110 nm)至与气态N2分子一致。形成的N2基团需克服24.0 kJ/mol的能垒和释放82.1 kJ/mol的能量才能从焦炭表面析出,N2析出后形成较为稳定的中间体IM4。最后,焦炭表面的C(O)以CO的形式解吸,生成产物P。该过程吸收了82.6 kJ/mol的能量,克服了427.6 kJ/mol的能垒,这与SENDT et al[45]的研究结果相一致。由图4所示的势能面来看,N2O—C反应是放热反应,在热力学上很容易发生。
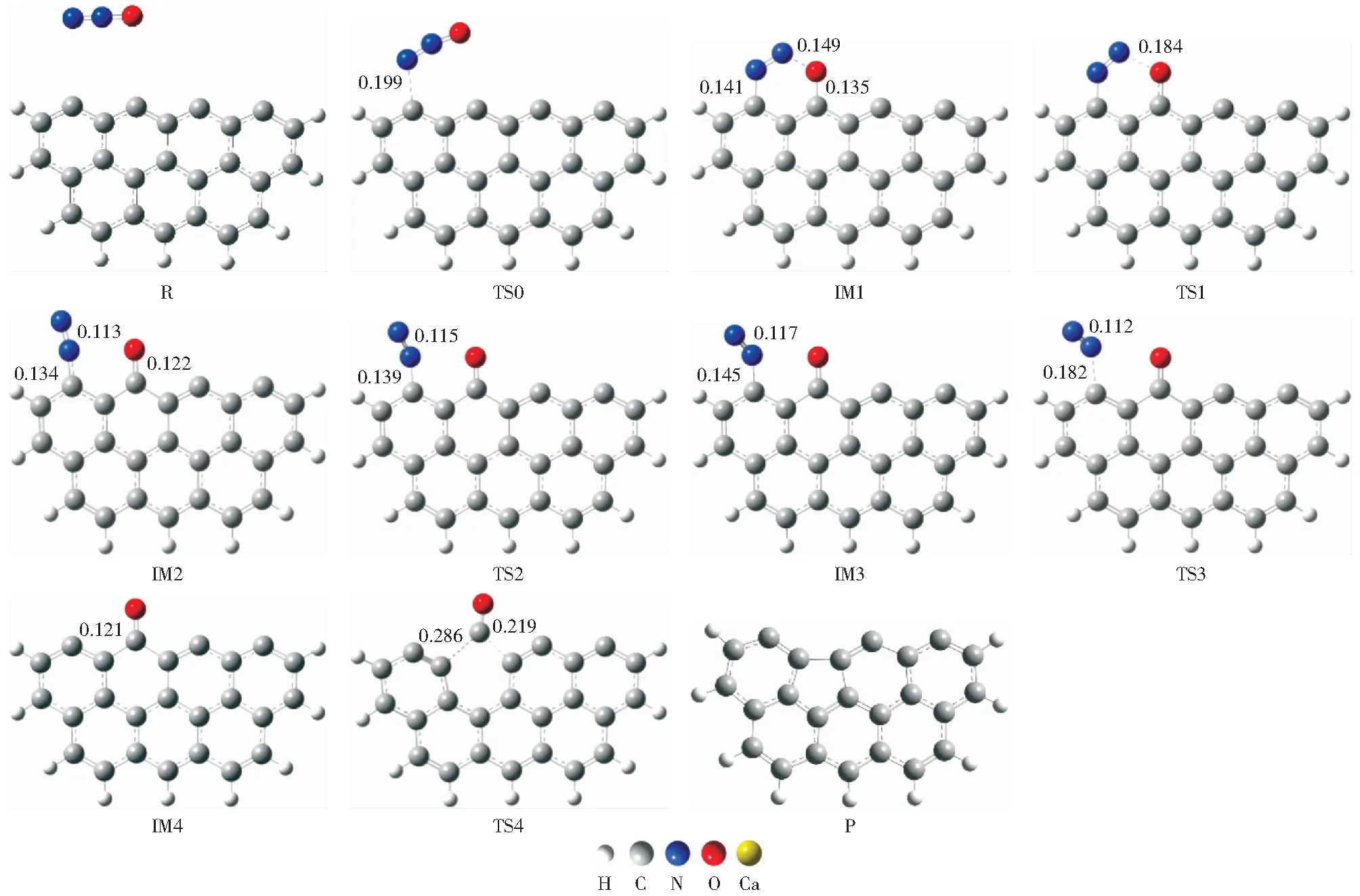
图3 原始Zigzag焦炭模型还原N2O的构型变化Fig.3 Configuration changes in chemisorption and reduction of N2O in pristine Zigzag char model
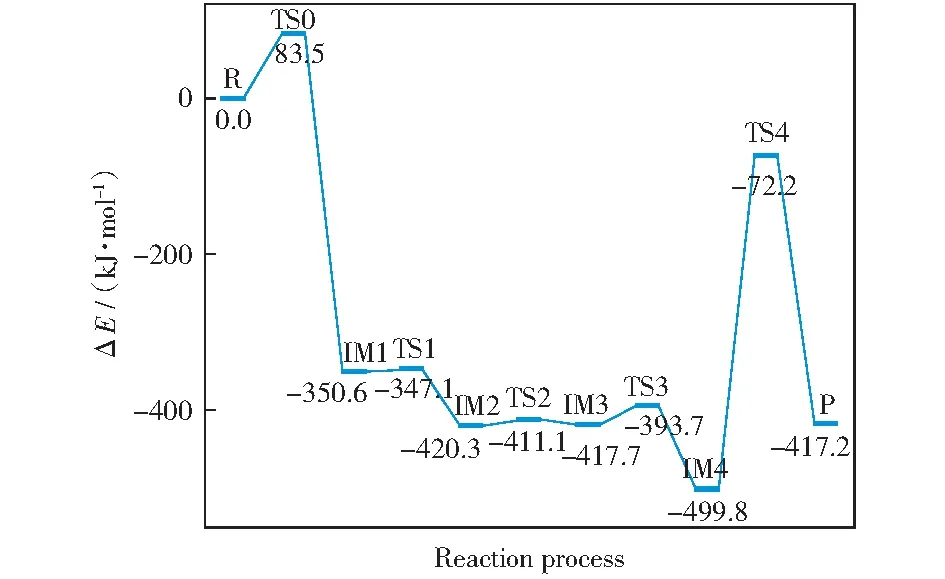
图4 原始Zigzag焦炭模型还原N2O的势能面Fig.4 Potential energy surface of chemisorption and reduction of N2O in pristine Zigzag char model
2.2 钙修饰的Zigzag焦炭模型还原N2O
Ca修饰的Zigzag焦炭模型还原N2O过程优化的几何构型和势能面分别如图5和图6所示。由图5可知,此过程共经历5个过渡态和4个中间体。首先,N2O以N—O侧向的形式吸附到焦炭表面,经历TS0生成中间体IM1。在此过程中,需要克服的能垒为76.8 kJ/mol,并释放出371.8 kJ/mol的能量。该步的能垒比原始Zigzag焦炭模型的能垒要小,且放热量更大,说明钙对焦炭表面吸附N2O有促进作用。这种促进作用可通过图7的静电势结果进一步说明。在图7中,红色和蓝色分别代表电子稀缺和电子富集,蓝色越深,表示这个位置聚集的电子越多,化学活性越高。由图7b可以看出,钙原子周围呈现红色,表示钙原子失去电子,焦炭边缘的碳原子带有更多的负电荷。N2O在焦炭表面的吸附是亲电反应,焦炭表面的活性位点带有的负电荷越多,越有利于N2O分子的吸附,因此,钙的修饰促进了N2O在焦炭边缘的吸附。由图6还可知,由IM1经历TS1到IM2需要克服的能垒仅为3.5 kJ/mol。IM2的能量比IM1的能量低84.4 kJ/mol,因此,前者更稳定。IM2→IM3的演化是一个放热过程(-7.7 kJ/mol)。在此过程中,C2—N10—N11的键角由173°(IM2)缩小到153°(TS2),再缩小到124°(IM3)。同时,N10—N11键长增加(0.113 nm→0.115 nm→0.117 nm),C2—N10键长也增加(0.134 nm→0.137 nm→0.146 nm)。下一步,N2从焦炭表面经历过渡态TS3解吸形成IM4,需要克服26.2 kJ/mol的能垒。该能垒低于原始Zigzag焦炭模型吸附还原N2O的计算值(39.9 kJ/mol)[22]。这进一步表明钙的修饰增加了焦炭表面的反应活性。最后,焦炭表面的C(O)以CO的形式从焦炭表面解吸,生成产物P。这一过程克服的能垒(400.1 kJ/mol)比原始Zigzag模型的能垒(427.6 kJ/mol)低,说明Ca的修饰降低了决速步的能垒,增加了反应速率,促进了N2O在焦炭表面的还原。因此,钙促进焦炭还原N2O的机理与文献[11]关于钙促进焦炭还原NO的机理相一致,都是对NOx还原产物和焦炭气化产物的解吸具有促进作用。由图6还可以看出,钙修饰的Zigzag模型还原N2O整体释放出436.7 kJ/mol的能量,这表明在热力学上是有利的。
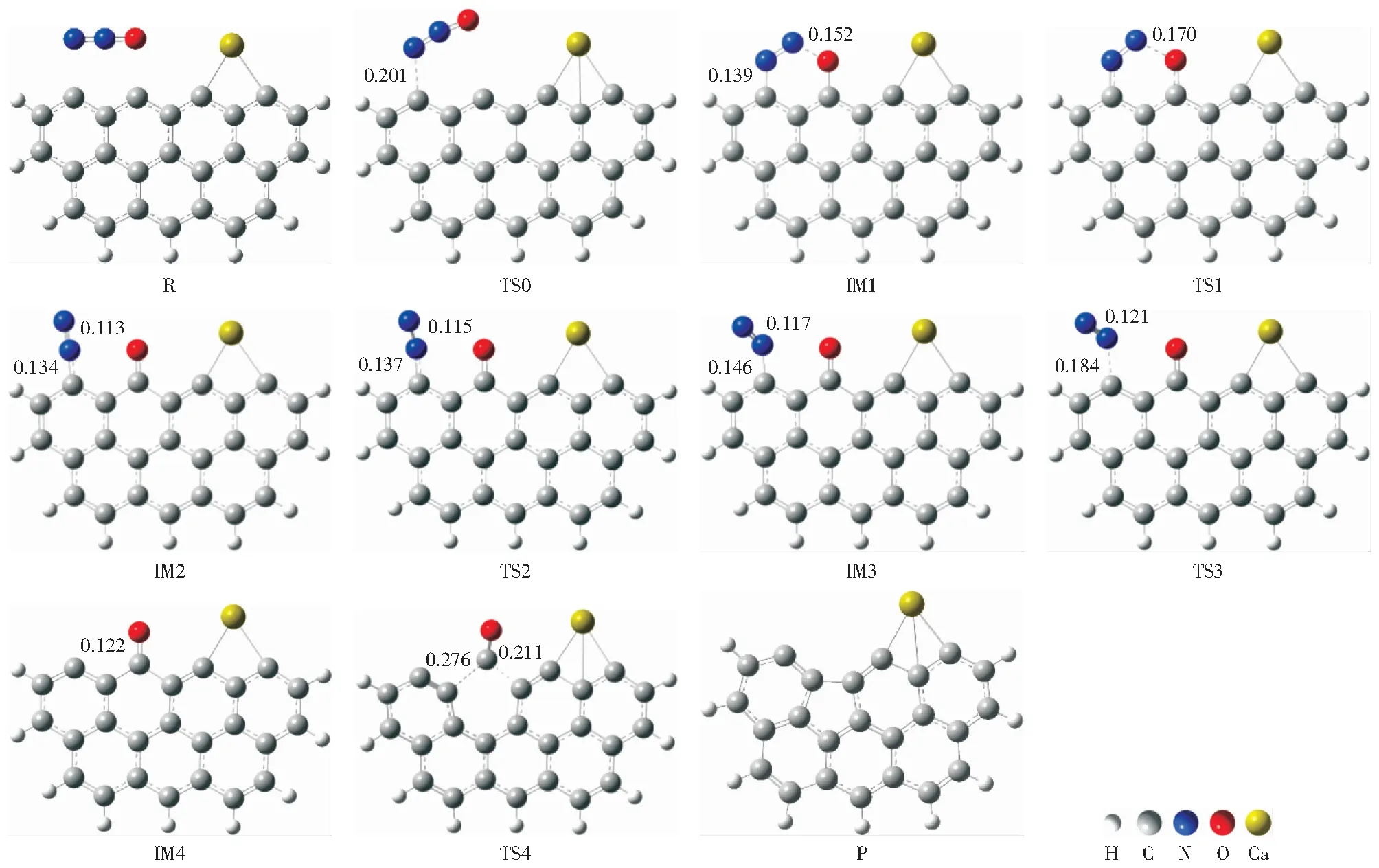
图5 钙修饰的Zigzag焦炭模型还原N2O的构型变化Fig.5 Configuration changes in chemisorption and reduction of N2O in Ca-decorated Zigzag char model

图6 钙修饰的Zigzag焦炭模型还原N2O的势能面Fig.6 Potential energy surface of chemisorption and reduction of N2O in Ca-decorated Zigzag char model
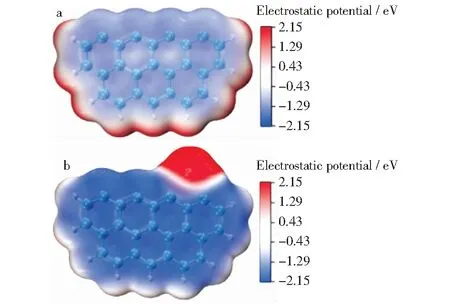
图7 钙修饰Zigzag焦炭模型前后的静电势比较Fig.7 Electrostatic potential of different modelsa—Pristine Zigzag char model;b—Ca-decorated Zigzag char model
利用Mayer键级可以确定分子结构中键的相对强度[46],定量描述重要化学键的形成和断裂,更好地理解原子水平的反应机制。通过计算每个单点结构(涉及IRC),确定每个结构中原子之间的Mayer键级,关键化学键的Mayer键级计算结果如图8所示。图8a表明了过渡态TS1的IRC路径中Mayer键级的变化。由图8a可以看出,N2O分子吸附到焦炭表面后N11与O12的键级逐渐降低到0,表明N11—O12键趋于分离。N10—N11键级有所增加,C2—N10的键级略有降低。TS2的IRC路径中Mayer键级的变化如图8b所示。由图8b可知,C2—N10的键级缓慢增加且保持稳定,说明该键不易断裂。同时,随着N10—N11键级增加,N10—N11键呈增强趋势。TS3的IRC路径中Mayer键级变化如图8c所示。由图8c可知,N10—N11的键级增加至2.7,显示出该键强度接近N≡N,N2的结构趋于稳定。C2—N10的键级逐渐降低,说明N2分子逐渐脱离C2位点,最后N2从焦炭边缘解吸出来。
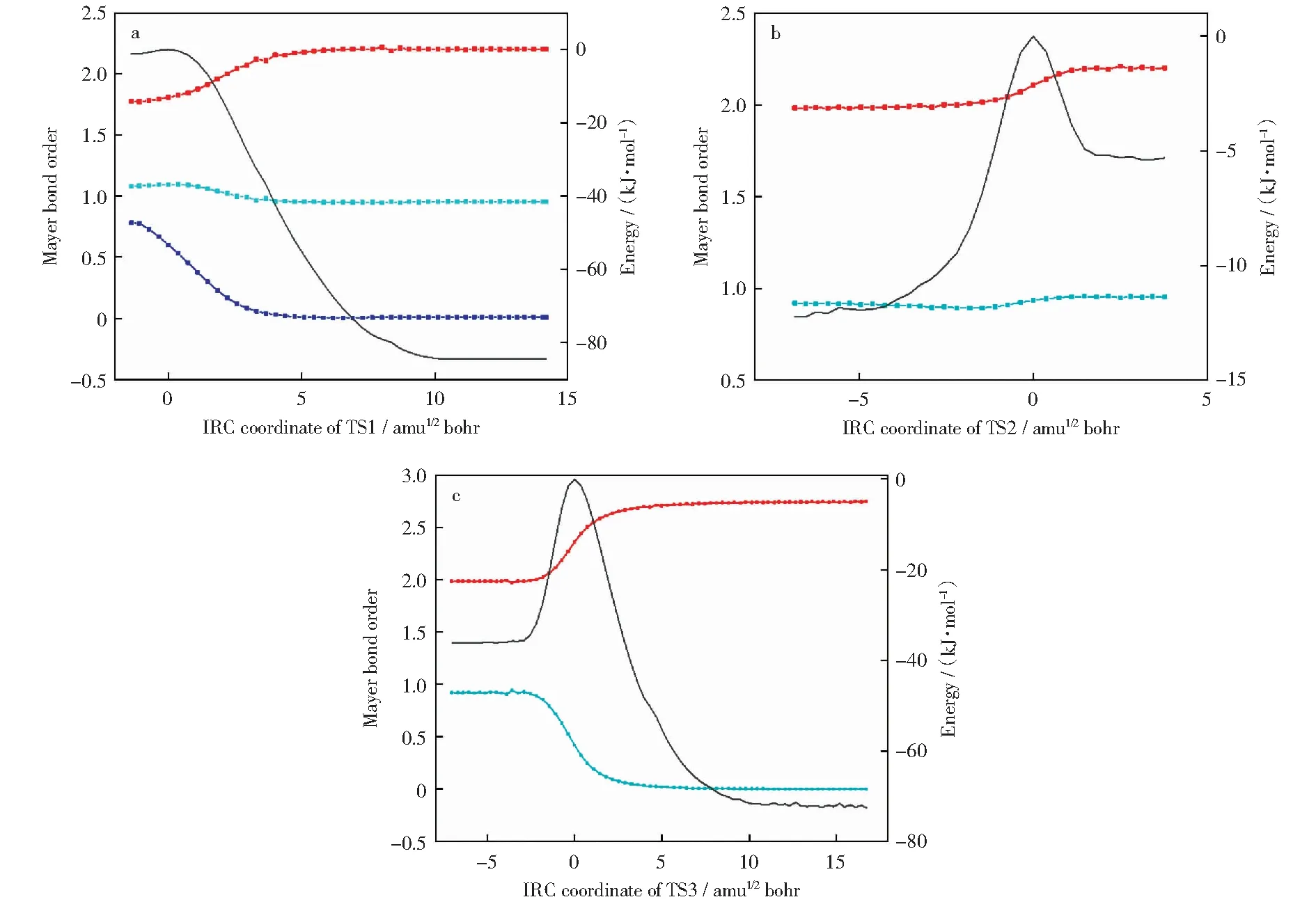
图8 Ca修饰的Zigzag焦炭模型还原N2O的过渡态IRC路径上的Mayer键级变化Fig.8 Mayer bond orders along IRC coordinate of TS for reduction of N2O in Ca-decorated Zigzag char model
2.3 羟基修饰的Zigzag焦炭模型还原N2O
羟基修饰的Zigzag焦炭还原N2O过程优化的几何构型和势能面分别如图9和图10所示。由图9和图10可知,此过程共经历10个过渡态(TS0~TS9)和9个中间体(IM1~IM9)。该反应路径从R到IM4的步骤与上文关于Ca修饰的焦炭模型的相应步骤基本相似,故这里不再赘述。N2解吸完成后,CO2在IM4开始解吸。C3—C4键断裂和C4—C6键形成,经历过渡态TS4形成中间体IM5,需要克服370.5 kJ/mol的能垒,吸收276.4 kJ/mol的能量。C4—C5键经历过渡态TS5断裂,形成C3—C5新键(键长为0.146 nm),促使中间体IM6形成,需要克服的能垒为47.9 kJ/mol,释放能量287.7 kJ/mol。该过程可通过IM5,TS5和IM6的Mulliken原子电荷进一步证实(见图11)。由图11可知,C3和C5在IM5上的原子电荷分别为0.070和0.002,在TS5上的原子电荷分别为0.024和-0.022。C3和C5之间的库仑引力增加促进了C3—C5键的形成。此外,在TS5中C4和C5的Mulliken原子电荷分别为0.317和-0.022,在IM6中它们的原子电荷分别为0.404和0.169。C4和C5之间的库仑斥力导致C4—C5键断裂。这些作用也导致了五元苯环的形成。由图10还可知,由于IM5和IM6之间的能垒很低,IM5不能作为稳定的中间产物存在,由于IM6的能量较低,它将保持稳定。H14通过TS6从O13转移到C9,需要克服292.7 kJ/mol的能垒。接下来,C4和O13原子通过TS7相互靠近,形成一个含有五元环的中间体IM8,需要克服13.5 kJ/mol的能垒,并释放25.4 kJ/mol的能量。对于过渡态TS8,C4—C6键断裂距离为0.272 nm,形成中间体IM9,需要跨越396.8 kJ/mol的能垒,吸收383.2 kJ/mol的能量,这一步是该路径的决速步。计算得到CO2的解吸能垒为396.8 kJ/mol,与SENDT et al[47]的实验结果(160 kJ/mol~440 kJ/mol)相一致。该能垒比原始Zigzag焦炭模型的能垒小,说明羟基修饰后促进了C(O)解吸,从而促进N2O在焦炭上的还原。羟基催化焦炭还原N2O的机理可以认为是羟基的O原子与N2wO的O原子以及焦炭的C原子结合形成CO2,从而完成C(O)的解吸。这与羟基催化焦炭还原NO的机理有所不同,ZHANG et al[26]发现羟基中的氧原子激活与其相连的C原子,C(O)配合物分解形成新的活性位点促进了另一个NO分子吸附。本研究中CO2解吸的能垒比CO解吸的能垒(427.6 kJ/mol)低,这与CHEN et al[21]的研究结果相一致。C9—O13键在过渡态TS9断裂,平衡距离为0.180 nm,对应的能垒为80.4 kJ/mol,释放85.4 kJ/mol的能量。最终,CO2被释放出来。由R→P过程的总能量变化可知,整个过程需要释放187.9 kJ/mol的能量。

图9 羟基修饰的Zigzag焦炭模型还原N2O的构型变化Fig.9 Configuration changes in chemisorption and reduction of N2O in OH-decorated Zigzag char model

图10 羟基修饰的Zigzag焦炭模型还原N2O的势能面Fig.10 Potential energy surface of chemisorption and reduction of N2O on OH-decorated Zigzag char
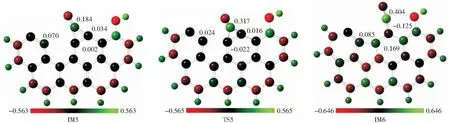
图11 IM5,TS5和IM6的Mulliken原子电荷Fig.11 Mulliken atomic charge of IM5, TS5 and IM6
2.4 动力学分析
根据过渡态理论(TST),活化能最高的基元反应是整个反应的决速步。由上述分析可知,原始焦炭模型及钙和羟基修饰的焦炭模型还原N2O反应的决速步分别为IM4→P,IM4→P和IM7→IM8。根据式(1)和式(2),在298.15 K~1 500.00 K温度范围内计算了三个模型还原N2O反应决速步的反应速率常数,结果如图12所示。此外,利用lnk对1 000/T进行线性拟合,得到活化能、指前因子等动力学参数,结果如表1所示。

表1 不同焦炭模型还原N2O反应的动力学参数Table 1 Fitted kinetic parameters of different Zigzag char models
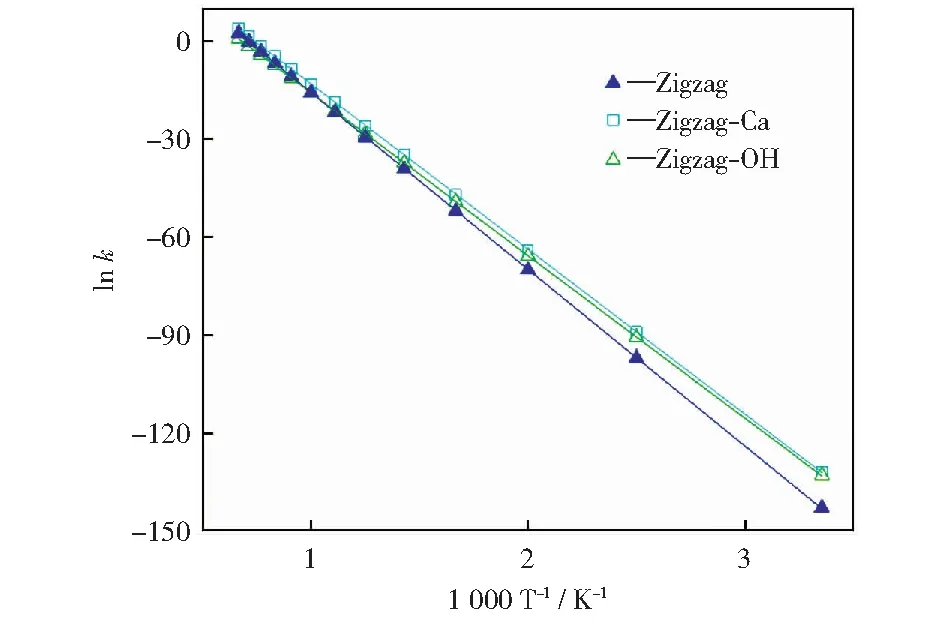
图12 不同Zigzag焦炭模型还原N2O决速步的反应速率常数Fig.12 Reaction rate constants for rate-limiting steps of reduction of N2O on different Zigzag char models
由图12可知,随着温度升高,反应速率常数均增大,说明温度升高对N2O-C反应有促进作用,这与实验研究结果相一致[48-49]。从lnk的斜率来看,温度对原始焦炭的影响大于对修饰后的焦炭的影响。钙和羟基修饰后的焦炭模型在低温下的反应速率大于原始焦炭模型在低温下的反应速率,而在高温下三种焦炭模型的反应速率差异很小。比较表1中的活化能可以看出,钙和羟基修饰后焦炭模型的活化能均低于原始焦炭模型的活化能,并且羟基的促进作用更强。
3 结 论
1) 钙修饰后焦炭还原N2O决速步的能垒降低了27.5 kJ/mol,表明钙修饰增加了焦炭表面的反应活性,未改变焦炭-N2O反应的产物,通过降低决速步的能垒增加了化学反应速率。
2) 羟基修饰焦炭模型前,N2O中的O原子与焦炭中的C原子结合形成CO,并最终解吸。羟基修饰焦炭模型后,羟基中的O原子与焦炭中的C原子以及N2O中的O原子结合形成CO2,反应过程中的中间体发生变化,解吸产物由CO转变为CO2,焦炭表面含氧配合物的解吸能垒降低,促进了焦炭对N2O的还原。
3) 钙修饰后N2O-C反应总体的放热量增加,而羟基修饰后焦炭还原N2O的总体放热量降低。钙和羟基对焦炭还原N2O都有促进作用,其中后者的作用更强。钙通过向焦炭转移电子使焦炭带有更多的负电荷,增加了焦炭边缘活性位点的反应活性,降低决速步能垒,增加反应速率。
猜你喜欢
世界农药(2023年8期)2023-09-04
燃料化学学报(2023年3期)2023-03-11
北京航空航天大学学报(2022年5期)2022-06-06
中国药学药品知识仓库(2022年10期)2022-05-29
大学化学(2021年8期)2021-09-26
中学课程辅导·教学研究(2021年8期)2021-07-14
燃料化学学报(2021年5期)2021-06-02
汕头大学学报(自然科学版)(2020年4期)2020-12-14
电脑知识与技术(2018年3期)2018-03-21
哈尔滨理工大学学报(2017年1期)2017-04-08
