
纳米球型Mo-MOF材料的调控合成及催化硫醚选择性氧化
2023-12-19 14:38郭昊天鲁新环孙凡棋陶艺元段金贵夏清华
高等学校化学学报 2023年12期
郭昊天,鲁新环,孙凡棋,陶艺元,段金贵,张 望,周 丹,夏清华
(1.湖北大学有机功能分子合成与应用教育部重点实验室,武汉 430062;2.南京工业大学材料化学工程国家重点实验室,南京 211816)
亚砜和砜类化合物由于在医药和农药等方面有广泛的应用[1,2],其需求量日益增长.亚砜和砜类化合物合成最直接的办法是硫醚的直接氧化,目前,用于硫醚氧化的氧化剂主要有氧气、过氧化氢及有机过氧化物、硝酸及硝酸盐和卤化物等[3,4].硝酸及硝酸盐的氧化性较强,但需要使用大量的硝酸,对环境的污染性较为严重.而卤化物虽然不会造成较重的环境污染,但是它对于硫醚氧化产物的选择性较差.目前研究中使用较多的是氧气和过氧化氢,过氧化氢作为一种绿色氧化剂[5],反应后只会产生水,但其稳定性较差且运输存在安全隐患,而氧气作为自然资源中较丰富的气体,成为硫醚氧化中更为绿色安全的氧化剂.然而,氧气分子的活化是较为困难的,这限制了它的使用,因此如何有效地活化氧气分子是一个重大挑战[6].过渡金属独特的电子结构能够活化氧分子,通常以Co,Cu,Fe,Mo和V为金属中心[7~11]与有机配体作用合成金属配合物.目前,以Mo为金属中心的催化剂催化氧气氧化反应的研究相对其它催化剂而言还较少[12~14].
金属有机框架(Metal organic frameworks,MOFs)[15~17]是一种由有机配体和无机金属离子配位组成的多孔材料,在催化[18~22]、分离[23]、气体储存[24]、传感器[25]和生物医学等领域有着广泛应用.2013年,Xuan等[26]报道了一种双吡啶基功能化手性Ti(salan)配体的合成,以制备多孔MOF,制备的材料在苯甲硫醚的过氧化氢氧化上表现出良好的催化活性,室温下搅拌72 h可以得到77%的氧化产物和81%的亚砜选择性.Granadeiro等[27]在2015年设计了一种锆有机骨架材料UiO-66,对于柴油中的有机硫化物的氧化有明显的催化性能,在过氧化氢存在下能够使96%及以上的硫化物氧化脱硫.Ye等[28]在2017年发现,通过部分钛离子交换可以提高UiO-66(Zr)的氧化脱硫(ODS)性能.由于催化活性位点Ti(Ⅳ)的加入,二苯并噻吩的氧化反应在有晶体缺陷的Ti-UiO-66催化下脱硫率高达91.7%.2019年,Ye等[29]发现使用硝基和氨基修饰UiO-66(Zr)也可以改变材料的催化活性,在不添加溶剂和调制剂的条件下,合成得到了UiO-66(Zr)-NO2和UiO-66(Zr)-NH3.此外,通过二苯并噻吩和4,6-二甲基二苯并噻吩的氧化脱硫反应,探究了官能团对UiO-66(Zr)催化活性的影响.Maksimchuk 等[30]在2021 年报道了两种新型Zr 有机骨架材料Zr-abtc 和MIP-200,分别是以3,3',5,5'-偶氮苯四甲酸(H4abtc)和5,5'-亚甲基二苯二甲酸为有机配体合成,前者在二苯并噻吩氧化脱硫上表现出的催化活性只稍弱于UiO-66.
从上述的研究中可以发现,对于MOFs 材料在硫醚氧化上的应用使用较多的氧化剂为过氧化氢,而空气/氧气的使用涉及较少.2021 年,Li 等[31]报道了一种以锕系金属铀为催化活性位点和1,1,2,2-四(4-羧基苯基)乙烯为配体合成得到的U-MOF材料.这种材料可以有效活化氧分子,使得苯甲硫醚在氧气气氛下选择性地转化为苯甲亚砜.然而,至今为止还没有以Mo为金属中心合成的金属有机骨架材料在硫醚类化合物氧气氧化上的报道.本文以价格相对较低的钼酸为钼源,使用简便易操作的静态混合溶剂热晶化法制备了Mo-MOF纳米球型催化剂材料,其相对其它催化剂具有更小的晶体结构和更分散的金属中心,对分子氧的活化有一定的促进作用.在未加其它助剂或还原剂的条件下,以氧气为氧化剂,在较低的反应温度下可以实现二苯基硫醚选择性氧化,且选择性形成了二苯基亚砜.
1 实验部分
1.1 试剂与仪器
对苯二甲酸(H2BDC,纯度>99.0%)、2-氨基对苯二甲酸,纯度>99.0%)、2,5-二羟基对苯二甲酸(纯度>99.0%),上海阿拉丁试剂公司;N,N-二甲基甲酰胺(DMF,纯度>98.5%)、无水乙醇(纯度99.5%)、氢氟酸(HF,纯度≥40.0%)、四氢呋喃(THF,纯度≥99.0%)、1,4-二氧六环(纯度>99.5%)、甲苯(纯度≥98.5%)、环己烷(纯度≥98.5%)、甲醇(纯度≥99.5%)、叔丁醇(纯度≥99.5%)、乙腈(纯度≥98.0%)、钼酸(纯度>85.0%)、二苯基硫醚(纯度≥98.0%)、苯甲硫醚(纯度≥99.0%)、二苄基硫醚(纯度≥99.0%)、二糠基硫醚(纯度≥98.0%)、糠基甲基硫醚(纯度≥97.0%)、二苯二硫醚(纯度≥98.0%)、二环己基二硫醚(纯度≥97.0%)、苯并噻吩(纯度≥97.0%)、二苯并噻吩(纯度≥98.0%)、4-甲氧基苯甲硫醚(纯度≥97.0%)、4-氯苯甲硫醚(纯度≥97.0%),麦克林试剂公司.
iChrom W5100型液相色谱仪(HPLC),大连依利特分析仪器公司;KLJX-47型均相反应器,烟台科立化工有限公司;D8型粉末X射线衍射仪(XRD),德国Bruker公司;DIGILAB Excalibur 型傅里叶变换红外光谱仪(FTIR),德国Bruker公司;JSM-6510A型场发射扫描电子显微镜(FESEM),日本电子公司;Chemstar TPX化学吸附仪(TPD),美国Quantachrome公司.
1.2 实验过程
1.2.1 催化剂的制备 静态溶剂热晶化法:将0.81 g H2MoO4(5 mmol)和0.83 g H2BDC(5 mmol)加入到盛有50 mL去离子水和无水乙醇(水/乙醇体积比为3∶7)的聚四氟乙烯内衬中,随后加入0.25 g 40% HF(5 mmol),在室温下搅拌1 h后,置于180 ℃烘箱中静态晶化20 h,取出冷却至室温,用去离子水抽滤洗涤3~4次,将所得固体在80 ℃下过夜烘干,得到样品记为Mo-MOF-180 ℃-20 h-Static.
动态溶剂热晶化法:称量的各物质质量和比例如上,将混合物在室温下搅拌1 h后,置于均相反应器中在180 ℃下动态晶化20 h,取出冷却至室温,用去离子水抽滤洗涤3~4次,将所得固体在80 ℃下过夜烘干,得到样品记为Mo-MOF-180 ℃-20 h-Dynamic.
静态干胶晶化法:称量的各物质质量和比例如上,将混合物在室温下搅拌1 h后,将内衬中的水分在烘箱中烘至约2 g,随后置于180 ℃烘箱中静态干胶晶化20 h,取出冷却至室温,用去离子水抽滤洗涤3~4次,将所得固体在80 ℃下过夜烘干,得到样品记为Mo-MOF-180 ℃-20 h-D-Static.
动态干胶晶化法:称量的各物质质量和比例如上,将混合物在室温下搅拌1 h后,将内衬中的水分在烘箱中烘至约2 g,随后置于均相反应器中在180 ℃下动态晶化20 h,取出冷却至室温,用去离子水抽滤洗涤3~4次,将所得固体在80 ℃下过夜烘干,得到样品记为Mo-MOF-180 ℃-20 h-D-Dynamic.
1.2.2 硫醚氧化反应过程 将20 mg催化剂、5.0 g溶剂和1.0 mmol底物二苯基硫醚加入50 mL不锈钢反应釜中,密封好后充入氧气至压强为1.0 MPa,置于配有加热套的磁力搅拌器上,以5 ℃/min的升温速率从室温升至一定反应温度.反应一段时间后,取下反应釜冷却至室温后,取反应液离心,分离反应液和催化剂,反应液使用HPLC(色谱柱型号为Hypersil ODS C18,250 mm×4.6 mm×5 μm.柱温箱温度为40 ℃,检测器为示差折光检测器,流动相为乙腈-水(体积比80∶20),流速为0.4 mL/min,进样量为10 μL)进行定量分析检测.
2 结果与讨论
2.1 催化剂的表征
图1(A)为不同合成方式合成的Mo-MOF材料的XRD谱图.可见,当采用静态晶化和动态晶化的合成方式时,材料的衍射峰较低,其中静态晶化合成的材料衍射峰较少,说明这种方式合成的材料的晶相较少.而使用静态干胶晶化和动态干胶晶化方式合成的材料的衍射峰数量较多,且强度较高,说明通过这两种合成方式得到的材料的晶相较为杂乱.图1(B)为在不同混合溶剂中合成的Mo-MOF材料的XRD谱图.可见,以DMF-甲醇、水-DMF和水-甲醇为混合溶剂分别合成的材料的衍射峰较高,说明这些材料的晶体尺寸较大.而当混合溶剂中加入乙醇时,材料的衍射峰强度明显下降.以甲醇-乙醇为混合溶剂合成的材料的衍射峰较多,且峰强度相差不大,说明这个材料的晶相较为复杂.而以DMF-乙醇为溶剂合成材料时,衍射峰数量明显下降,在2θ=5°~10°和2θ=10°处存在2个衍射峰,说明材料可能有两种不同的晶体结构.当混合溶剂为水-乙醇时,材料只在2θ=5°~10°处存在衍射峰,说明以此种混合溶剂合成材料时形成的晶体结构较为接近,因此表现为一个宽峰.图1(C)为不同水-乙醇比例下合成的Mo-MOF材料的XRD谱图.可见,当溶剂中乙醇的比例逐渐增加时,材料衍射峰的数量先减少后增多,当水-乙醇体积比为3∶7时合成的材料的晶相较为统一,而乙醇比例较低或过高会导致合成的材料的晶相较为复杂.图1(D)为以水和乙醇为混合溶剂,不同晶化温度合成的Mo-MOF材料的XRD谱图.可见,在晶化温度只有120 ℃时,材料的衍射峰强度较高.当晶化温度升高到150 ℃后,几个主要的衍射峰的强度明显减弱,在2θ=5°~10°范围内出现一个新的衍射峰,表明材料的晶体结构发生了改变.当晶化温度为180 ℃时,材料的衍射峰只有2θ=5°~10°范围内的峰,说明材料的晶体结构出现了明显改变.随着晶化温度进一步升高到200 ℃,其XRD谱图中出现了新的小衍射峰.

Fig.1 XRD patterns of Mo-MOF(180 ℃-20 h) catalysts synthesized by different methods(A),Mo-MOF(180 ℃-20 h-Static) catalysts synthesized in different mixed solvents(B) Mo-MOF(180 ℃-20 h-Static) catalysts synthesized in solvents at different H2O/C2H5OH volume ratios(C) and Mo-MOF(20 h-Static) catalysts synthesized under different crystallization temperatures(D)
图2为不同混合溶剂中合成的Mo-MOF材料的SEM照片.从图2(A)可以看到,水-乙醇为混合溶剂合成的Mo-MOF 材料的形貌为较完整的球形结构,晶体之间较为分散,有利于催化活性位点的暴露.使用水-甲醇为混合溶剂合成的材料的团聚性较为严重,表现为较大的无规则块状结构[图2(B)].在图2(C)中显示的是以水-DMF 为混合溶剂合成的材料的形貌,整体上为较大的堆积的块状晶体结构.以DMF-甲醇为混合溶剂合成的材料的形貌显示在图2(F)中,可以看到,这个条件下形成的晶体为大小不一的块状结构.从图2(D)和(E)可以看到,当在溶剂中加入乙醇后,合成的材料中存在少量的球形晶体,但大部分的晶体仍为大小不一的块状结构.虽然材料的晶体尺寸大大减小,但整体上的形貌仍较为杂乱.因此,选择水和乙醇为最优的混合溶剂.

Fig.2 SEM images of Mo-MOF(180 ℃-20 h-S) catalysts synthesized in different mixed solvents
图3为不同水-乙醇比例合成的Mo-MOF材料的SEM照片.当溶剂体积比为7∶3时[图3(A)],材料的晶体结构为较小的无规则的块状.当溶剂比例调整为3∶7 时[图3(B)],材料整体为完整的球形结构.进一步增加溶剂中乙醇的比例时[图3(C)],晶体的球形结构变得畸形且出现团聚现象.当只以乙醇为溶剂时[图3(D)],材料的晶体结构较为杂乱,存在球形和块状结构.这说明当合成溶剂中乙醇的比例较低或过高时都不利于材料球形晶体结构的生成,故选择水-乙醇体积比为3∶7为最优的溶剂比例.

Fig.3 SEM images of Mo-MOF(180 ℃-20 h-Static) catalysts synthesized in mixed solvents at different H2O/C2H5OH volume ratios
图4 是水和乙醇(体积比为3∶7)为溶剂,不同晶化温度合成的Mo-MOF 材料的SEM 照片.从图4(A)中可以看到,当晶化温度为120 ℃时,材料为大的不规则块状晶体结构.当晶化温度为150 ℃时合成的材料的晶体结构较为杂乱,存在球形、棒状及块状晶体[图4(B)].如图4(C)所示,随着晶化温度上升到180 ℃,材料主要呈现为球形,生成的催化位点可以很好地暴露出来,因此材料的催化活性较高.当进一步升高晶化温度至200 ℃时[图4(D)],材料的球形结构部分发生了破裂,且有较多尺寸较小的片状晶体出现.这说明当晶化温度较低时,晶体不能生长为球形结构,而温度过高时晶体生成的球形结构不稳定.因此,选择180 ℃为最优的晶化温度.为了进一步探究所制得材料的球形结构,对材料进行了FESEM表征.

Fig.4 SEM images of Mo-MOF(20 h-Static) catalysts synthesized at different crystallization temperatures
由FESEM 照片[图5(A)~(D)]可见,材料的晶体尺寸在1 μm 左右,且晶体是由纳米粒子堆积形成,表面凹凸不平,这也使得材料暴露出更多的催化活性位点,因此材料的催化活性较高.图5(E)为场发射扫描电子显微镜照片(FESEM).由Mo-MOF-180 ℃-20 h-Static 的能量色散X 射线能谱(EDX)可见,C,O和Mo元素的能谱峰较为明显,说明合成的催化剂中含有这3种元素[图5(F)].从材料的元素面分布图中可见,C,O和Mo元素在晶体表面分散均匀,且具有良好的空间重叠[图5(G)~(I)].因此选择水-乙醇体积比为3∶7为最优的溶剂比例.

Fig.5 FESEM images(A—E),EDX(F) and mapping analysis(G—I) of Mo-MOF(180 ℃-20 h-Static) catalysts
图6(A)为不同晶化温度合成的Mo-MOF材料的NH3-TPD谱图.可以看到,当晶化温度较低时,材料的酸强度较低,随着晶化温度的升高材料的酸性增强,当晶化温度过高时,材料的酸强度反而降低.SEM表征的结果表明,在温度较低时晶体生长为较大的块状,能够暴露的酸性位点较少,温度过高时晶体的结构发生改变,导致了酸性位点数量降低.图6(B)为不同晶化温度合成的Mo-MOF材料的吡啶红外光谱图.可以看到,不同温度下合成的材料在1549,1491和1454 cm-1处有相同的吸收峰,其中,1549 cm-1处的吸收峰可归属于吡啶中的N原子与B酸位点连接后的特征峰,1454 cm-1为L酸位点与N原子连接后的特征峰,1491 cm-1则是B+L酸的吸收峰[32,33].当晶化温度为150 ℃,B酸的吸收峰强度较弱,而L酸的吸收峰强度相对较强.在晶化温度为180 ℃时L酸的吸收峰强度最高,进一步升高温度后峰强度降低,这说明在晶化温度较高时材料倾向于生成L酸位点,而温度过高会使晶体结构出现破损,进而导致一些L酸位点的缺失,因此材料的催化活性降低.
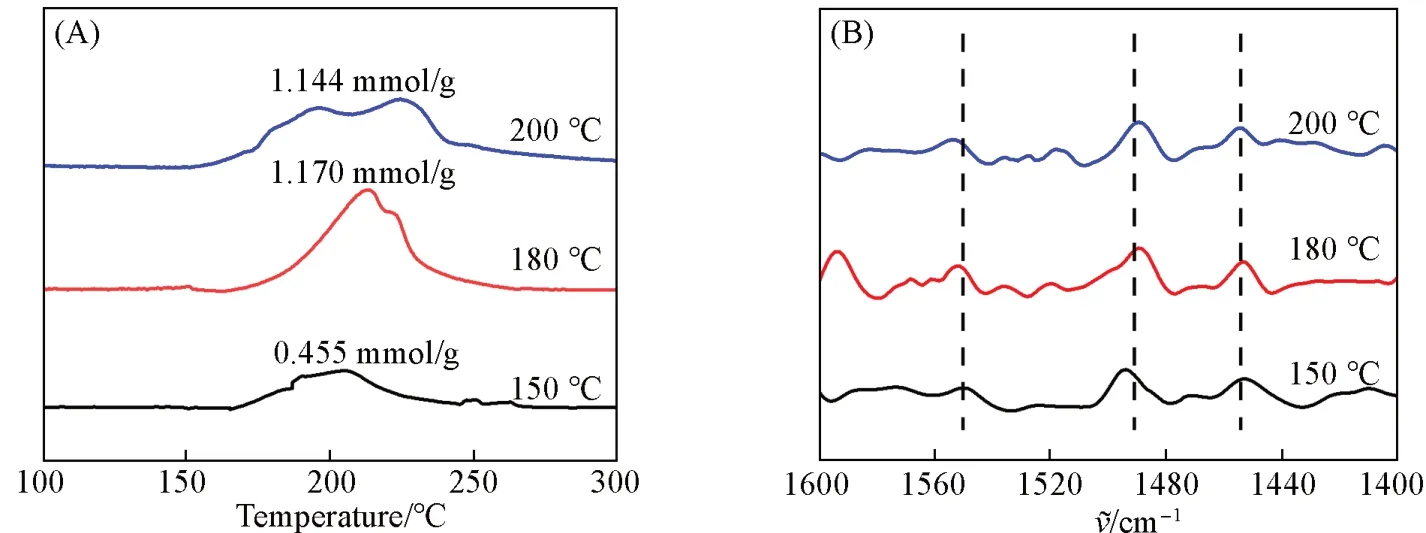
Fig.6 NH3-TPD profiles(A) and Py-IR spectra(B) of Mo-MOF(20 h-Static) catalysts synthesized at different crystallization temperatures
2.2 催化硫醚选择性氧化
图7(A)显示了合成方式对Mo-MOF催化活性的影响.当使用静态溶剂热法合成Mo-MOF时,二苯基硫醚的转化率为100%,产物二苯基亚砜的选择性为86.4%.当使用动态溶剂热法合成材料时,转化率下降至51.2%,催化剂的活性有了明显下降.静态干胶晶化法和动态干胶晶化法合成的材料对于二苯基硫醚氧化的催化活性较低,分别表现为2.0%和8.1%的转化率,说明干胶法不利于合成Mo-MOF催化材料.图7(B)显示了配体种类对Mo-MOF催化活性的影响.当使用对苯二甲酸作为配体时,材料的催化活性较强,能够得到100%的转化率和86.4%的二苯基亚砜选择性.使用2-氨基对苯二甲酸作为配体时,二苯基硫醚的转化率和选择氧化产物二苯基亚砜的选择性均降低,说明使用这种配体合成的材料的催化活性较低.当使用2,5-二羟基对苯二甲酸作为配体时,二苯基硫醚几乎没有转化,说明以这种配体合成的Mo-MOF的几乎没有催化活性.
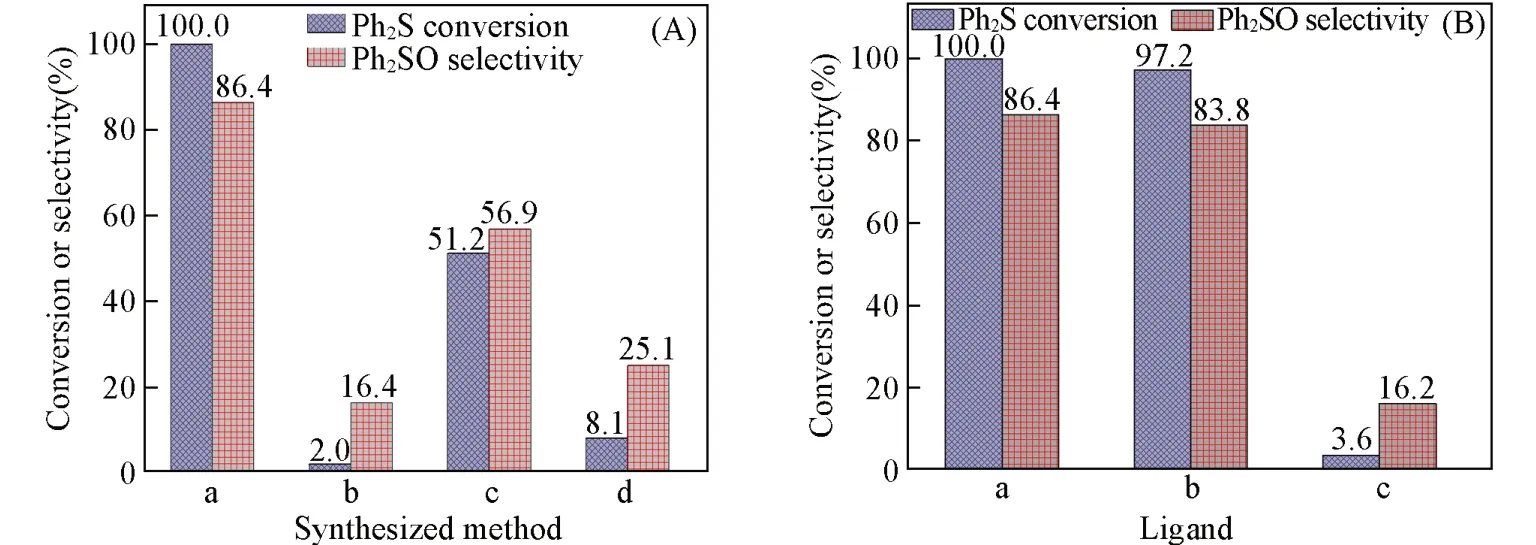
Fig.7 Effects of synthesized methods(A) and ligands(B) on the catalytic activity of Mo-MOF(180 ℃-20 h)
图8(A)显示了混合溶剂组成对Mo-MOF催化活性的影响.当使用水-乙醇为混合溶剂合成Mo-MOF材料时,二苯基硫醚的转化率为100%,且二苯基亚砜的选择性有86.4%.而使用水-甲醇为混合溶剂时,转化率有了明显下降.结合材料的SEM照片可以发现,以水-甲醇为混合溶剂合成的材料团聚较为严重,暴露出的活性位点较少,所以材料的催化活性明显降低.以水-DMF、甲醇-乙醇、DMF-乙醇和DMF-甲醇为混合溶剂合成的材料其催化活性都较低,这也与其无法暴露出较多的活性位点有关.图8(B)显示了水-乙醇体积比对Mo-MOF催化活性的影响.随着混合溶剂中水含量的降低,二苯基硫醚的转化率呈现出先增高后降低的趋势.当水-乙醇的比例为7∶3时,转化率可以达到65.8%.减少溶剂中水的含量后,在水-乙醇的体积比为3∶7时,转化率为100%,且二苯基亚砜的选择性为86.4%.当仅以乙醇为溶剂合成材料时,转化率降低至10.8%.结合SEM 照片可以发现,水-乙醇的体积比为3∶7后,材料表现为规整的球形结构,暴露出更多的活性位点,所以其催化活性更高,而以乙醇为溶剂合成的材料晶体较小且团聚性严重,所以表现出的催化活性较低.
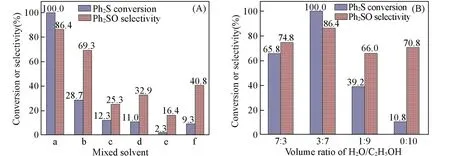
Fig.8 Effects of synthesized solvents(A) and volume ratio of H2O/C2H5OH(B) on the catalytic activity of Mo-MOF(180 ℃-20 h-Static)
图9(A)显示了晶化温度对Mo-MOF催化活性的影响.当晶化温度为120 ℃时,二苯基硫醚的转化率仅为10.2%.随着晶化温度的升高,转化率先提高后降低,在180 ℃时达到最高,为100%.结合SEM照片可以看到,当晶化温度较低时,晶体生长成较大的块状,无法形成较多的球形结构,暴露的催化活性位点较少.晶化温度升高后,形成了较多的球形晶体,催化活性位点暴露较多,因此催化活性较高.当晶化温度过高时,晶体结构破损,晶体表面的活性位点减少,材料的催化活性降低.图9(B)显示了晶化时间对Mo-MOF 催化活性的影响.当晶化时间为10 h时,二苯基硫醚的转化率只有35.5%,二苯基亚砜的选择性为68.5%.晶化时间延长至20 h 时,转化率显著增高至100%,选择性也达到了86.4%.随着晶化时间的进一步延长,转化率下降而选择性增加,但二苯基亚砜的产率下降.
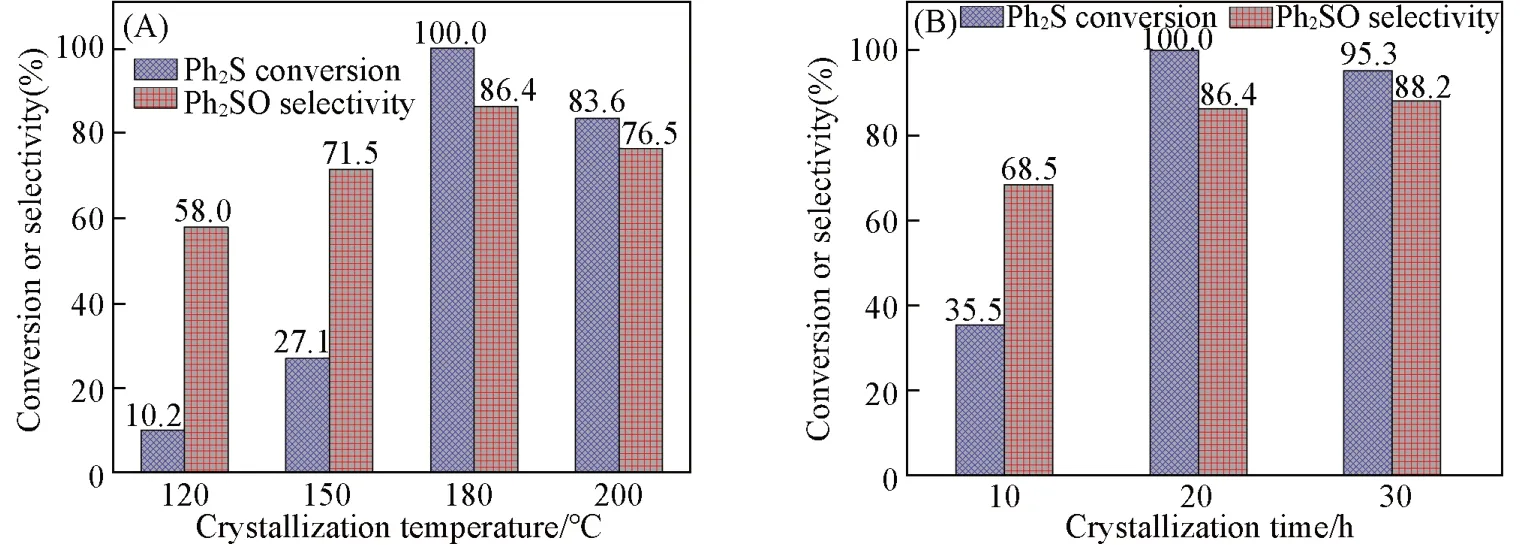
Fig.9 Effects of different crystallization temperatures(A) and crystallization times(B) on the catalytic activity of Mo-MOF-Static
图10(A)显示了不同反应温度对硫醚氧化的影响.当反应温度在90 ℃以下时,二苯基硫醚的反应活性较低,转化率较低,在80 ℃时仅有22.7%.当反应温度上升至90 ℃时,二苯基硫醚的反应活性较高,能够得到100%的转化率以及86.4%的亚砜选择性.反应温度继续升高后,转化率不变而选择性下降,这是因为温度较高时亚砜进一步转化成了砜.图10(B)显示了反应时间对硫醚氧化的影响.从图中可以看到,当反应温度适宜时,在反应时间仅有0.5 h 时,二苯基硫醚的转化率已经达到了91.1%,在反应时间延长至0.75 h后,底物已经全部转化且有86.4%的亚砜产物.随着反应时间的进一步延长,亚砜逐渐转化为砜,因此亚砜的选择性逐渐降低.
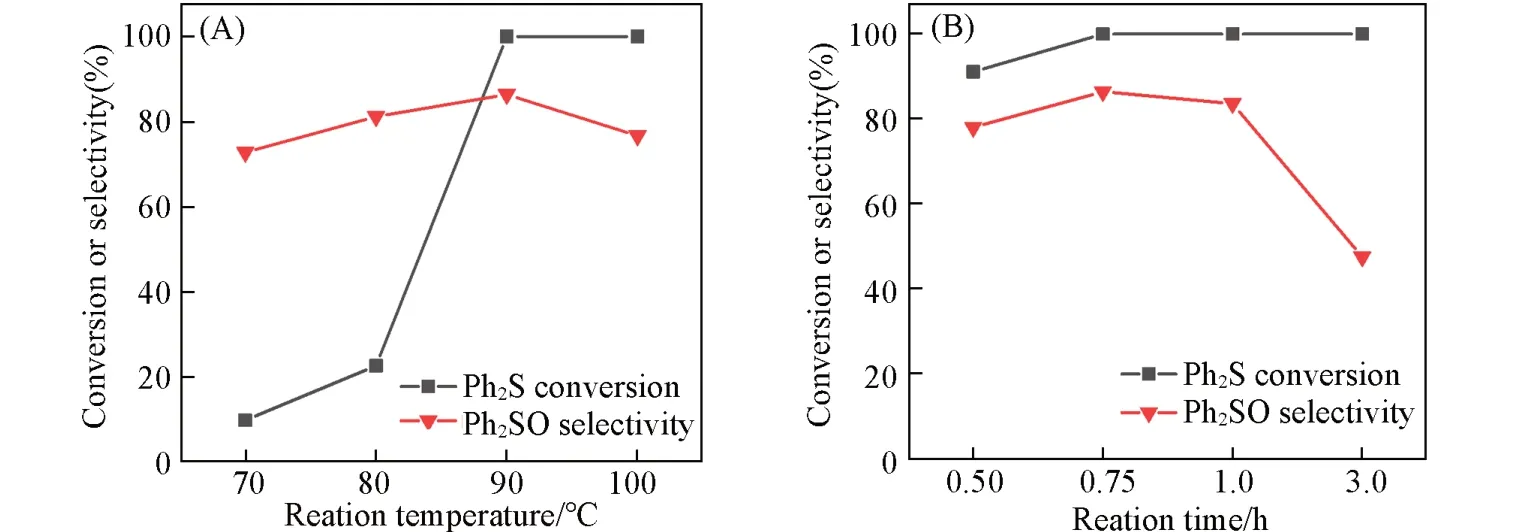
Fig.10 Effects of different reaction temperature(A) and reaction time(B) on the oxidation of thioethers
综上所述,本文以钼酸为金属源、对苯二甲酸为配体,以水-乙醇(体积比为3∶7)为混合溶剂在温度为180 ℃下静态溶剂热晶化20 h,合成了纳米球型Mo-MOF材料.最优条件制备的Mo-MOF催化剂在无其它助剂下可实现二苯基硫醚高效转化为选择氧化产物,产物中二苯基亚砜的选择性可达到86.4%.其良好的催化性能归因于催化材料的球型晶体表面高度分散的活性金属Mo.材料的合成方法、有机配体的种类、合成溶剂的组成及比例等影响因素在控制硫醚选择性氧化反应中起了重要作用.
猜你喜欢
山西化工(2023年10期)2023-11-15
水产科学(2023年4期)2023-07-22
山东化工(2019年7期)2019-04-27
中国资源综合利用(2017年1期)2018-01-22
浙江化工(2017年11期)2017-12-19
中国塑料(2017年2期)2017-05-17
百科知识(2016年18期)2016-10-28
浙江化工(2015年10期)2015-03-10
苏州科技大学学报(工程技术版)(2015年3期)2015-02-28
食品科学(2013年10期)2013-12-23
