
电化学炔烃氧化断键构建α-羰基芳酰胺衍生物的研究
2024-01-04 23:59杨帆高梦杨桂春
湖北大学学报(自然科学版) 2024年1期
杨帆,高梦,杨桂春
(湖北大学化学化工学院,湖北 武汉 430062)
0 引言
碳碳三键的选择性断裂反应可以实现对有机分子骨架的直接修饰,也被认为是新一代物质转化的途径[1]。但是,碳碳三键键能相对较高,其断裂需要强氧化剂,高温等苛刻的反应条件,反应过程中伴随着大量副产物的产生[2-4]。近年来,报道了一系列新型的过渡金属催化的炔烃选择性断键的研究,以简单炔烃快速合成酰胺、酸、酯、腈等化合物,极大的丰富了炔烃参与的有机合成方法学[5-8]。然而,这些方法也存在着重金属残留、催化剂昂贵难以制备等问题。寻找不依赖强氧化剂和昂贵金属催化剂的温和条件实现碳碳三键的选择性断裂依旧是有机合成领域的研究热点。
α-羰基酰胺衍生物是一类重要的胺类化合物,具有良好的生物活性,广泛存在于天然产物中,是重要的有机合成中间体,并且被用于药物分子结构设计中。治疗湿疹的药物Tacrolimus(FK506)、治疗丙型肝炎的新药Telaprevir和Boceprevir均含有α-羰基酰胺结构单元[9-10]。报道的合成α-羰基酰胺方法大多以重金属催化、碘催化为主,存在催化剂价格昂贵、污染环境等问题[11-13],寻找便宜、高效、绿色、环保的合成方法一直是有机合成工作者追求的目标。
近年来,电化学有机合成取得了令人瞩目的进展,引起了广泛的关注[14]。相较于传统的合成方法,电化学合成利用电子作为绿色的反应试剂,无需添加额外的氧化剂或还原剂,也不依赖于昂贵的过渡金属催化剂[15]。此外,电化学合成通过对电极电势的精准调控使其能活化传统手段难以活化的反应底物,从而能在相对温和的室温条件下快速高效的构建目标分子,同时避免了大量副产物产生[16]。本研究基于电化学合成的优势,以1,4-二苯基-2-丁炔-1,4-二酮为起始原料,与亚硝基苯发生自由基加成反应,随后在氧气的帮助下发生分子内的重排反应选择性的实现碳碳三键的断裂,在室温下一步简便高效合成具有高附加值的α-羰基酰胺衍生物。该方法操作简单,反应条件温和,无需金属催化剂和外加氧化剂的使用,符合绿色化学的发展需求。
1 实验部分
1.1 试剂和仪器试剂:亚硝基苯,1,4-二苯基-2-丁炔-1,4-二酮,30%过氧化氢水溶液(H2O2),叔丁基过氧化氢(TBHP),四丁基碘化铵(n-Bu4NI),四丁基六氟磷酸铵(n-Bu4NPF6),四丁基氟化铵(n-Bu4NF·H2O),二氯甲烷(CH2Cl2),二氯乙烷(DCE),乙腈(CH3CN)等均为分析纯,无需纯化直接使用。
仪器:电解仪器为双显示恒电位器(DJS-292 B) (中国),阳极为碳棒电极(Φ6 mm),阴极为铂片电极(15 mm×15 mm×0.5 mm)。
1.2 底物的合成依据文献报道的方法合成亚硝基苯[17]。
氮气气氛下,将苯胺(20 mmol)溶解于50 mL二氯甲烷溶液中,将Oxone (22 mmol, 13.5 g) 缓慢加入50 mL水中,逐滴加入到苯胺溶液中。薄层色谱(TLC)监测反应过程,直到苯胺完全消耗。反应完毕,加入水,有机层用乙酸乙酯萃取,分液,收集有机相,分离得到的有机层依次用饱和NaHCO3溶液、少量蒸馏水、饱和食盐水进行洗涤,分液,有机层用无水硫酸钠干燥,过滤收集滤液,浓缩除去溶剂,通过柱层析分离(V环己烷∶V乙酸乙酯= 100∶1),得到黄色固体粉末,产率为45%。
1.3 实验方法在氧气和室温下将1a(0.3 mmol),2a(0.15 mmol),四丁基碘化铵(0.3 mmol)加到25 mL一体池中,并加入CH2Cl2(6 mL)溶解,碳棒为阳极,铂片为阴极,10 mA恒流电解6 h,薄层色谱和液相色谱-质谱法跟踪分析,硅胶柱层析(V环己烷∶V乙酸乙酯= 10∶1)得到产物3a(产率:45%,16 mg)。
2 结果与讨论
2.1 电化学合成α-羰基酰胺衍生物的条件优化以碳棒作为阳极,铂片为阴极的一体池中,使用亚硝基苯(1a)和1,4-二苯基-2-丁炔-1,4-二酮(2a)作为模板底物(图1),探讨反应条件对合成α-羰基酰胺(3a)的影响。
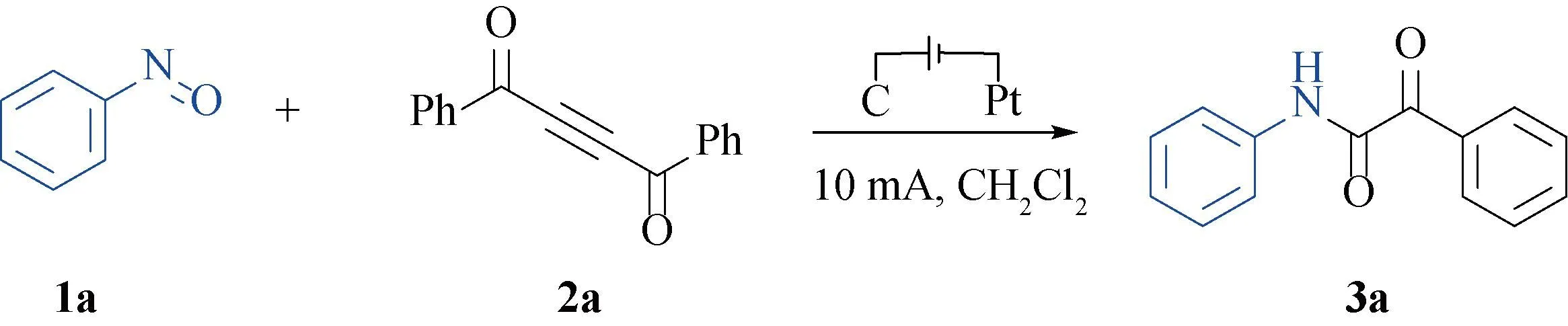
图1 电化学合成α-羰基酰胺
在室温和O2氛围下,将亚硝基苯1a(0.3 mmol),1,4-二苯基-2-丁炔-1,4-二酮2a(0.15 mmol),n-Bu4NI(0.3 mmol)及6 mL CH2Cl2加入到一体池中,电极为C(+)/Pt(-),10 mA恒流电解6 h,获得了45%的3a(表1序号1)。当使用H2O2代替O2球作为氧化剂时,3a产率为40%(表 1序号2)。当使用TBHP代替O2球作为氧化剂时,3a产率为43%(表 1序号3)。当使用反应氛围为N2时,反应受到抑制,得到微量的3a(表 1序号4)。这说明该反应的进行需要氧气的参与。当使用n-Bu4NF·H2O和n-Bu4NPF6代替n-Bu4NI作为电解质时,3a的产率下降明显,产率分别为27%和12%(表 1序号5和6)。随后,我们探索了不同溶剂对反应的影响,发现把溶剂换成乙腈和二氯乙烷后,3a的产率也有明显的降低,产率分别为19%和26%(表 1序号7和8)。在不通电的条件下,反应无法进行(表1序号10)。

表1 电化学合成α-羰基芳酰胺的反应条件优化结果
2.2 底物普适性研究在确定最优反应条件后,对该电化学合成α-羰基芳酰胺衍生物反应的底物范围进行了普适性研究,其结果如图2。
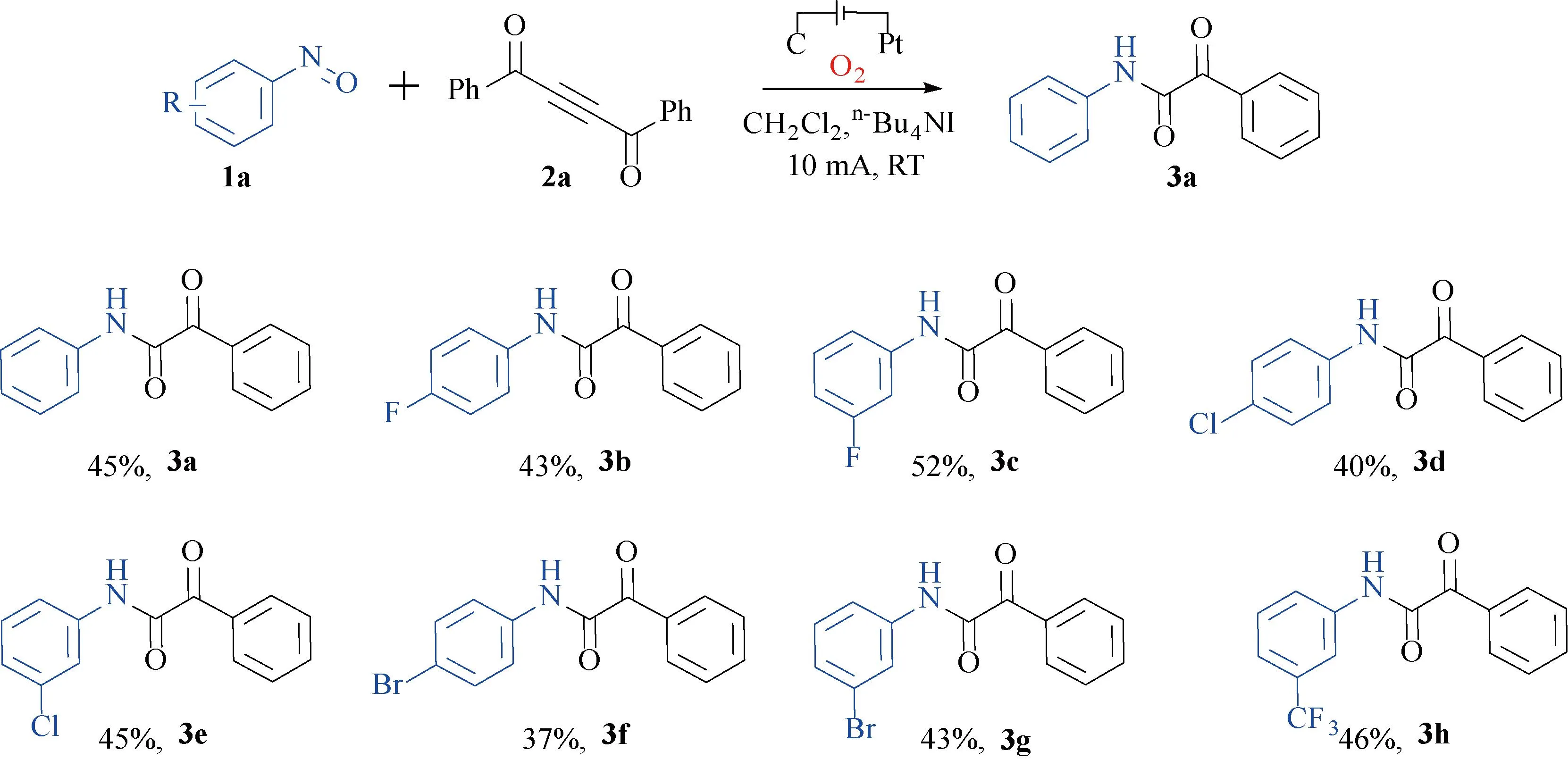
a标准反应条件:1 (0.3 mmol),2 (0.15 mmol),n-Bu4NI (0.3 mmol),碳棒,铂片,氧气球,一体池,室温,反应6 h
由图2可知:在室温,O2氛围下,反应电极为C/Pt,电流为10 mA,溶剂为CH2Cl2,电解质为n-Bu4NI,反应6 h时。研究了一系列具有吸电子基团(—F,—Cl,—Br,—CF3)及不同取代位点的底物,其中,含-F取代的底物3b和3c,对应产率分别为43%,52%。此外,含-Cl取代的底物3d,3e,其对应产率为40%,45%。当取代基为-Br,获得了化合物3f,3g,3f产率为37%,3g为43%。当取代基为CF3基团,如3h,产率为46%。这些数据表明该方法具有一定的普适性,值得注意的是,当吸电子基位于间位时,较对位具有相对较高的产率,且产率与取代基的吸电子强度呈正相关趋势。通过X-射线技术确认产物3a的结构。
3 目标产物的图谱表征
(3a) 2-diphenyl,2-oxo-nacetamide:1HNMR(400 MHz, Chloroform-d)δ9.00 (s, 1H), 8.40 (d,J=7.39 Hz, 2H), 7.70 (d,J= 8.0 Hz, 2H), 7.64 (t,J= 7.39 Hz, 1H), 7.49 (t,J= 7.8 Hz, 2H), 7.39 (t,J= 7.9 Hz, 2H), 7.20 (t,J= 7.4 Hz, 1H)。13CNMR(101 MHz, Chloroform-d)δ159.06, 136.75, 134.71, 77.26~77.06 (m), 131.53, 77.58~77.38 (m), 129.30, 128.64, 125.38, 76.94~76.74 (m), 120.06。HRMS(ESI-MS)m/zcalcfor [3a+H]+(C14H12NO2): 226.086 3, found: 226.085 9。
(3b) N-(4-fluorophenyl)-2-oxo-2-phenylacetamide:1HNMR(400 MHz, Chloroform-d)δ9.02 (s, 1H), 8.39 (d,J= 7.8 Hz, 2H), 7.66 (td,J= 10.2, 9.7, 6.0 Hz, 3H), 7.49 (t,J= 7.7 Hz, 2H), 7.07 (t,J= 8.5 Hz, 2H)。13CNMR(101 MHz, Chloroform-d)δ187.40, 161.10, 158.92, 158.66, 134.75, 133.00, 132.77 (d,J= 2.9 Hz), 131.46, 128.61, 121.74 (d,J= 8.0 Hz), 115.97 (d,J= 22.6 Hz)。HRMS(ESI-MS)m/zcalcfor [3b+H]+(C14H11FNO2): 244.076 8, found: 244.076 6。
(3c) N-(3-fluorophenyl)-2-oxo-2-phenylacetamide1HNMR(400 MHz, Chloroform-d)δ9.12 (s, 1H), 8.35 (d,J= 7.6 Hz, 2H), 7.63 (dd,J= 19.0, 9.3 Hz, 3H), 7.46 (t,J= 7.5 Hz, 3H), 7.39~7.27 (m, 3H), 6.86 (t,J= 7.5 Hz, 2H)。13CNMR(101 MHz, Chloroform-d)δ187.15, 164.25, 161.81, 159.09, 138.22 (d,J= 10.9 Hz), 134.84, 132.91, 131.50, 130.38 (d,J= 9.1 Hz), 128.65, 115.47 (d,J= 3.1 Hz), 112.19, 111.98, 107.66, 107.40, 77.48, 77.16, 76.84。HRMS(ESI-MS)m/zcalcfor [3c+H]+(C14H11FNO2): 244.076 8, found: 244.076 6。
(3d) N-(4-chlorophenyl)-2-oxo-2-phenylacetamide1HNMR(400 MHz, Chloroform-d)δ9.02 (s, 1H), 8.39 (d,J=7.9 Hz, 2H), 7.66 (d,J= 8.7 Hz, 3H), 7.50 (t,J= 7.6 Hz, 2H), 7.35 (d,J=8.6 Hz, 2H)。13CNMR(101 MHz, Chloroform-d)δ187.20, 158.94, 135.35, 134.90, 133.04, 131.59, 130.50, 129.40, 128.73, 121.27, 77.48, 77.16, 76.84。HRMS(ESI-MS)m/zcalcfor [3d+H]+(C14H11ClNO2): 260.047 3, found: 260.046 9。
(3e) N- (3-chlorophenyl) -2-oxo-2-phenylacetamide1HNMR(400 MHz, Chloroform-d)δ9.04 (s, 1H), 8.39 (d,J= 7.6 Hz, 2H), 7.85 (s, 1H), 7.66 (t,J= 7.4 Hz, 1H), 7.57~7.46 (m, 4H), 7.30 (t,J= 8.1 Hz, 1H), 7.16 (d,J= 8.0 Hz, 1H)。13CNMR(101 MHz, Chloroform-d)δ187.01, 158.92, 137.79, 134.90 (d,J=7.8 Hz), 132.86, 131.52, 130.23, 128.65, 125.38, 120.07, 117.98。HRMS(ESI-MS)m/zcalcfor [3e+H]+(C14H11ClNO2): 260.047 3, found: 260.046 9。
(3f) N- (4-bromophenyl) -2-oxo-2-phenylacetamide1HNMR(400 MHz, Chloroform-d)δ9.01 (s, 1H), 8.39 (d,J= 8.0 Hz, 2H), 7.66 (t,J= 7.39 Hz, 1H), 7.59 (d,J= 8.7 Hz, 2H), 7.49 (t,J= 7.7 Hz, 4H)。13CNMR(101 MHz, Chloroform-d)δ187.07, 158.84, 135.74, 134.83, 132.91, 132.26, 131.50, 128.64, 121.48, 118.09。HRMS(ESI-MS)m/zcalcfor [3f+H]+(C14H11BrNO2): 303.996 8, found: 303.996 5。
(3g) 2-oxo-2-phenyl-N-(3-bromophenyl) acetamide1HNMR(400 MHz, Chloroform-d)δ9.07 (s, 1H), 8.36 (d,J= 7.7 Hz, 2H), 7.97 (s, 1H), 7.63 (t,J= 7.4 Hz, 1H), 7.58 (d,J= 8.0 Hz, 1H), 7.48 (t,J= 7.7 Hz, 2H), 7.29 (d,J= 8.0 Hz, 1H), 7.21 (t,J= 8.0 Hz, 1H)。13CNMR(101 MHz, Chloroform-d)δ187.07, 159.00, 137.98, 134.89, 77.26~77.06 (m), 132.89, 131.54, 77.58~77.38 (m), 130.53, 128.68, 118.53, 76.94~76.75 (m), 128.32, 122.93 (d,J= 7.1 Hz)。HRMS(ESI-MS)m/zcalcfor [3g+H]+(C14H11BrNO2): 303.996 8, found: 303.996 5。
(3h) 2-oxo-2-phenyl-N-(3-(trifluoromethyl) phenyl) acetamide1HNMR(400 MHz, Chloroform-d)δ9.27 (s, 1H), 8.37 (d,J= 7.6 Hz, 2H), 8.07 (s, 1H), 7.87 (d,J= 7.8 Hz, 1H), 7.64 (t,J= 7.4 Hz, 1H), 7.46 (dt,J= 15.0, 7.7 Hz, 4H)。13CNMR(101 MHz, Chloroform-d)δ187.10, 159.25, 137.35, 134.97, 132.87, 77.26~77.06 (m), 131.83, 131.53, 77.58~77.38 (m), 129.83, 128.71, 125.21, 76.94~76.75 (m), 123.12, 122.50, 121.88 (q,J= 3.7 Hz), 116.87 (q,J= 3.8 Hz)。HRMS(ESI-MS)m/zcalcfor [3h+H]+(C15H11F3NO2): 294.073 6, found: 294.073 3。
4 机理验证
为了研究该反应可能的机理,进行控制实验(图3所示)。在氧气和室温的条件下,将苯胺(0.3 mmol),1,4-二苯基-2-丁炔-1,4-二酮2a(0.15 mmol),四丁基碘化铵(0.3 mmol)加到25 mL一体池中并加入CH2Cl2(6 mL)溶解,碳棒为阳极,铂片为阴极,10 mA恒流电解6 h,未得到产物3a,而是得到加成产物。这表明,亚硝基苯在该反应中并未还原生成苯胺参与反应。
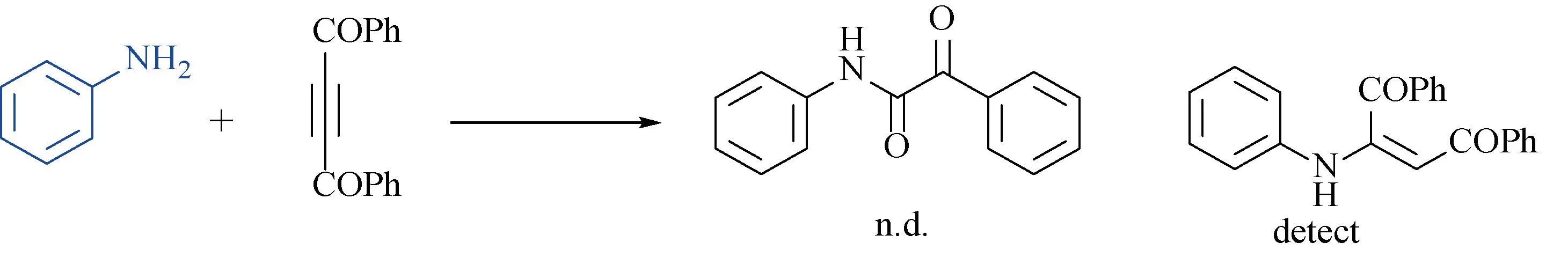
图3 控制实验
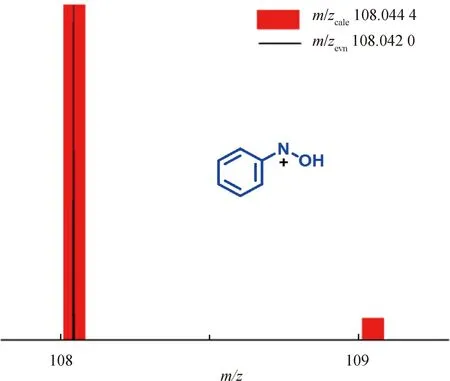
图4 质谱跟踪检测到的反应中间体片段及其拟合
为了进一步揭示该反应的机理,采用质谱进行了跟踪反应。在反应时长为5 min,10 min,15 min,30 min,1 h,2 h,3 h,4 h,5 h进行微量取样,用于质谱检测,检测到关键的反应中间体片段(图5)。
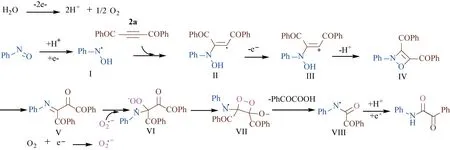
图5 反应机理推测
根据文献[18]和实验结果,推测了一个可能的反应机理(图5)。首先,亚硝基苯在阴极被还原得到亚硝基自由基I,2a与之发生自由基加成得到中间体II。中间体II发生单电子氧化进一步环化得到N-O四元环中间体IV,中间体IV经过重排生成中间体V,另一方面,O2在阴极还原得到超氧自由基负,超氧自由基负与中间体V加成得到中间体VI,随后经过分子内四元环状过渡态重排断键得到VIII,VIII进一步还原质子化得到目标产物3a。
5 结论
本研究以亚硝基苯和1,4-二苯基-2-丁炔-1,4-二酮为原料,通过电化学单电子还原的策略选择性的还原亚硝基苯产生亚硝基自由基,进而对1,4-二苯基-2-丁炔-1,4-二酮进行自由基加成,随后在氧气的作用下,通过分子内重排实现了碳碳三键的断裂,室温下成功构建出了一系列α-羰基芳酰胺。该方法利用“电子”作为绿色氧化剂,反应条件简单,温和,产物分离纯化方便,是一种高效、经济、环保的有机合成方法,对碳碳三键的断裂以及α-羰基酰胺的构建有着重要的研究意义。
猜你喜欢
陶瓷学报(2021年5期)2021-11-22
山东化工(2019年7期)2019-04-27
中国资源综合利用(2017年1期)2018-01-22
中国塑料(2017年2期)2017-05-17
苏州科技大学学报(工程技术版)(2015年3期)2015-02-28
应用化工(2014年1期)2014-08-16
应用化工(2014年1期)2014-08-16
应用化工(2014年4期)2014-08-16
中国氯碱(2014年10期)2014-02-28
生物加工过程(2013年1期)2013-03-11
