
小檗碱衍生物的合成及抗菌活性研究*
2024-01-16 07:20万升标
中国海洋大学学报(自然科学版) 2024年2期
葛 莲, 程 珊, 吕 钦, 万升标**
(1. 中国海洋大学医药学院,海洋药物教育部重点实验室, 山东 青岛 266003;2. 青岛海洋科学与技术试点国家实验室 海洋药物与生物制品功能实验室, 山东 青岛 266237)
耐药性细菌严重威胁着全球人民的生命健康,在众多耐药菌株中,耐甲氧西林金黄色葡萄球菌(Methicillin-ResistantStaphylococcusaureus,MRSA)和耐万古霉素肠球菌(Vancomycin-Resistant Enterococci,VRE)危害程度尤为突出[1]。仅在2014年,因MRSA引起的肺部感染夺走了全球150万人的生命,其中大多数死亡病例发生在发展中国家[2]。所以,基于新的抗菌药物靶点开发出新型抗菌药物是科学界亟待解决的问题之一。
细胞分裂温度敏感体Z(Filamenting temperature-sensitive mutant Z,FtsZ)是近年来研究的一个热门抗菌靶点,具有三磷酸鸟苷(Guanosine triphosphate,GTP)酶活性,与原核细胞的分裂过程密切相关[3]。FtsZ是由两个结构域组成的保守结构,其一是N端GTP酶区域,另一个是C端GTP酶激活区域。N端GTP酶区域的核苷酸结合位点与C端GTP酶激活区域的T7环在连接处结合形成GTP酶活性位点,通过中心的长螺旋H7相链接形成小分子结合口袋[4]。FtsZ蛋白几乎对所有细菌的生长都十分关键并且其形态上高度保守不易变异,这就为开发FtsZ抑制剂以抵抗多药耐药创造了可能[5-6]。当FtsZ活性受到抑制时,细菌无法完成增殖分裂形成狭长丝状或肿大球状结构,进而导致细菌死亡[7-8]。
已有不少研究者致力于开发FtsZ抑制剂,现有的FtsZ抑制剂大体上可以分为两类:天然产物类和合成类[9]。小檗碱是从中药黄连中提取出来的异喹啉生物碱,能够以大肠杆菌FtsZ蛋白为靶点,破坏FtsZ原丝的稳定性,并抑制FtsZ蛋白的GTP酶活性,是天然产物类FtsZ抑制剂(见图1)。小檗碱在医学界被用作抗菌剂,具有广谱抗菌活性,对革兰氏阳性菌和革兰氏阴性菌都有抑制作用,但抗菌活性不强,最低抑菌浓度在64 μg·mL-1以上[10]。3-甲氧基苯甲酰胺是一种合成类的FtsZ抑制剂,虽然抑菌活性不佳(对枯草芽孢杆菌的最小抑菌浓度为4 000 μg·mL-1),但具有优异的靶向活性,能靶向枯草芽孢杆菌中的FtsZ蛋白,诱导细胞丝状化并最终导致细胞裂解死亡,为新型FtsZ抑制剂的开发奠定了良好的基础。Haydon等[11]在3-甲氧基苯甲酰胺的基础上对甲氧基进行结构修饰,最后从500个3-甲氧基苯甲酰胺衍生物中筛选出了化合物PC190723,经过抗菌活性测试发现其最小抑菌浓度为0.5~1 μg·mL-1,抗菌效果要比3-甲氧基苯甲酰胺高出数千倍。之后又将PC190723的噻唑并吡啶改成苯并噻唑,发现MIC值由1.0 μg·mL-1降为0.25 μg·mL-1,抗菌活性有所提高[12]。基于上述研究基础对小檗碱进行结构改造,将苯并噻唑类活性片段与小檗碱杂合,以期望提高小檗碱的抗菌活性(见图2)。
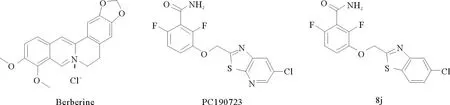
图1 部分FtsZ 抑制剂及其衍生物的化学结构
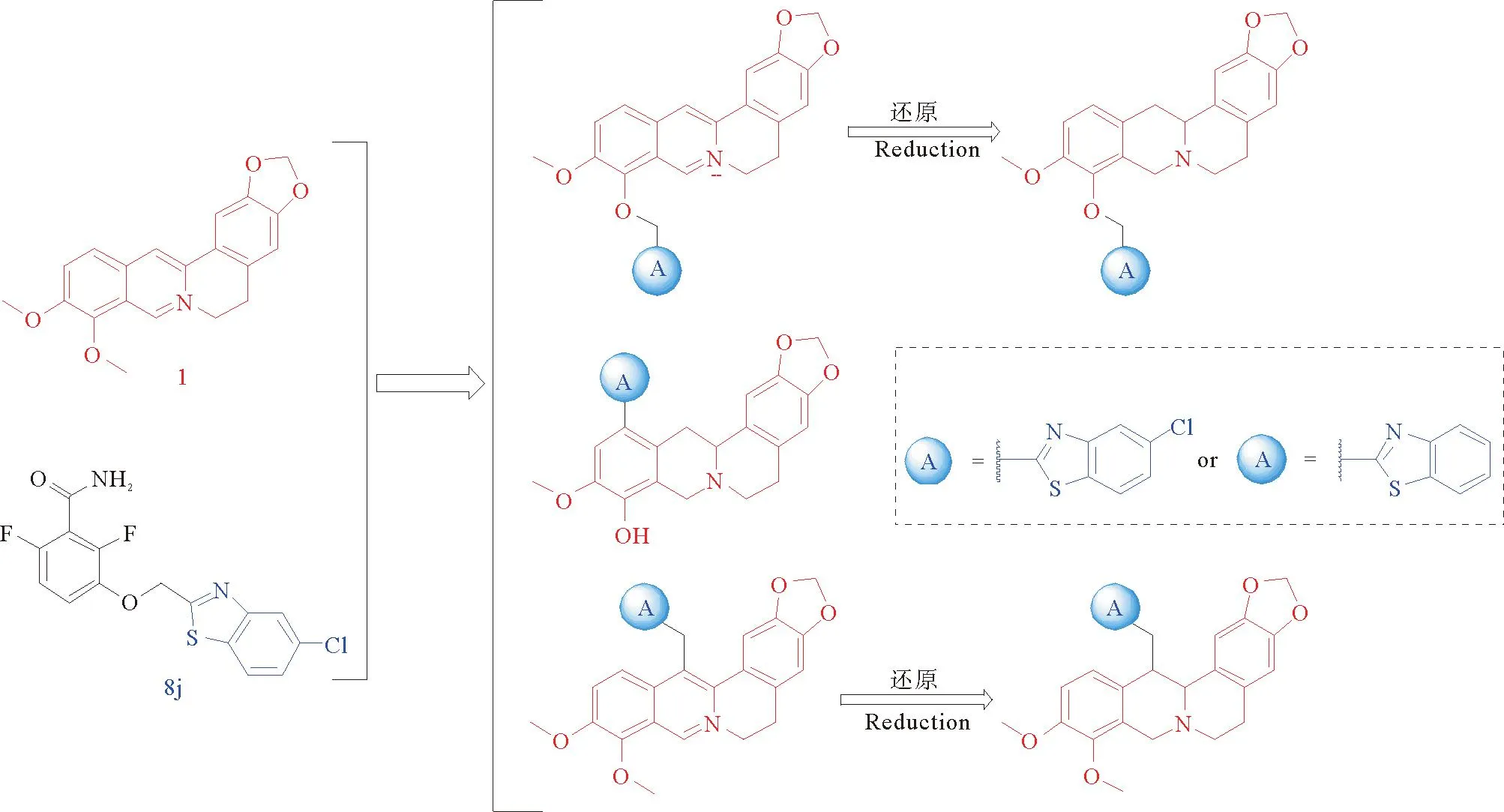
图2 小檗碱衍生物的设计
1 实验材料
1.1 主要仪器
JEOLJNM-EPC 400 NMR核磁共振仪( 捷欧路科贸有限公司);Agilent DD2 500 MHz 核磁共振仪(安捷伦科技有限公司);Q-TOF Ultima GLOBAL 质谱仪(英国Micromass公司);Merck 25 TLC 硅胶板(硅胶 60 GF254,0.25 mm,德国 Merck 公司);柱层析硅胶(200~300 目,青岛美高集团有限公司)。
1.2 化学试剂
盐酸小檗碱水合物(批号 E2211028,上海阿拉丁生化科技股份有限公司),2-氨基-4-氯-苯硫酚(批号 G2108067,上海阿拉丁生化科技股份有限公司),2-氨基苯硫酚(批号 L2103389,上海阿拉丁生化科技股份有限公司),碘化钠(批号 L2116064,上海阿拉丁生化科技股份有限公司),六亚甲基四胺(批号 X210628A,上海麦克林生化科技有限公司)。反应溶剂均为烟台远东精细化工有限公司分析纯试剂。
1.3 实验菌株
金黄色葡萄球菌(S.aureusATCC 6538)、耐甲氧西林金黄色葡萄球菌(MRSA ATCC 43300)、铜绿假单胞菌(P.aeruginosaATCC 10145)和大肠杆菌(E.coliATCC 11775)四种菌株均购自美国菌种保藏中心。
2 化合物的合成
2.1 9-(5-氯苯并噻唑-2-甲氧基) 小檗碱(5a)和9-(苯并噻唑-2-甲氧基) 小檗碱(5b)的合成
2.1.1 小檗红碱(2) 将小檗碱盐酸水合物1(5 000 mg, 13.47 mmol)于190 ℃下加热搅拌1 h,得到紫红色固体粗产物,产率90%。1H NMR (400 MHz, DMSO-d6) δ 9.86 (s, 1H), 7.96 (d,J=10.7 Hz, 1H), 7.89 (s, 1H), 7.74 (s, 1H), 7.59 (s, 2H), 7.04 (s, 1H), 6.14 (s, 2H), 4.68 (t, 2H), 4.02 (s, 3H), 3.08 (t, 2H)。
2.1.2 5-氯-2-(氯甲基)苯并噻唑(4a)和2-(氯甲基)苯并噻唑(4b) 将2-氨基-4-氯苯硫酚(3 200 mg, 20 mmol)或2-氨基苯硫酚(2 500 mg, 20 mmol)溶解于甲苯(50 mL)中,再向反应体系中逐滴加入氯乙酰氯(4 520 mg, 40 mmol)和三乙胺(2 020 mg, 20 mmol),于室温下反应20 min,之后加热到105 ℃反应3 h。用薄层色谱监测反应完全,用饱和碳酸氢钠溶液淬灭反应,盐水洗涤,乙酸乙酯萃取三次。将三次萃取得到的有机相收集起来并用无水硫酸钠干燥,之后将溶剂旋干。用石油醚/乙酸乙酯(v/v=20∶1)进行柱层析得到白色固体,产率56%。(4a) MS(ESI):m/z[M+H]+218.10; (4b) MS(ESI):m/z[M+H]+184.65。
2.1.3 9-(5-氯苯并噻唑-2-甲氧基) 小檗碱(5a)和9-(苯并噻唑-2-甲氧基) 小檗碱(5b) 将小檗红碱2(322 mg, 1 mmol)和化合物4a(1 085 mg, 5 mmol)或者化合物4b(915 mg, 5 mmol)溶解于乙腈(15 mL)中,再向反应体系中加入碘化钠(750 mg, 5 mmol)和碳酸钾(690 mg, 5 mmol),加热回流反应5 h。反应结束后,过滤除去固体残渣,再将滤液旋干。用二氯甲烷/甲醇(v/v=100∶1)进行柱层析得到黄色固体,产率30%。(5a)1H NMR (400 MHz, DMSO-d6) δ 9.97 (s, 1H), 8.98 (s, 1H), 8.25 (dd,J=11.5, 8.9 Hz, 2H), 8.15 (d,J=2.1 Hz, 1H), 8.08 (d,J=9.1 Hz, 1H), 7.81 (s, 1H), 7.57 (dd,J=8.6, 2.1 Hz, 1H), 7.10 (s, 1H), 6.19 (s, 2H), 5.78 (s, 2H), 4.94 (t,J=6.3 Hz, 2H), 4.08 (s, 3H), 3.22 (t,J=6.3 Hz, 2H);13C NMR (101 MHz, DMSO-d6) δ 171.74, 149.85, 146.05, 128.52, 127.93, 127.87, 125.71, 124.81, 122.97, 122.62, 110.98, 108.53, 105.61, 100.90, 71.62, 59.63, 55.94, 54.21, 51.48, 36.47, 29.81, 29.63; MS(ESI):m/z[M+H]+504.09. (5b)1H NMR (400 MHz, DMSO-d6) δ 9.96 (s, 1H), 8.97 (s, 1H), 8.26 (d,J=9.2 Hz, 1H), 8.22~8.16 (m, 1H), 8.10~8.03 (m, 2H), 7.80 (s, 1H), 7.62~7.44 (m, 2H), 7.10 (s, 1H), 6.18 (s, 2H), 5.80 (s, 2H), 4.94 (t,J=6.3 Hz, 2H), 4.09 (s, 3H), 3.21 (t,J=6.3 Hz, 2H);13C NMR (101 MHz, DMSO-d6) δ 169.51, 153.02, 149.93, 146.24, 146.03, 143.38, 135.24, 130.79, 128.59, 127.90, 126.17, 125.19, 124.72, 123.15, 121.90, 111.01, 108.53, 105.62, 100.89, 71.73, 59.63, 55.97, 54.23, 51.46, 36.48, 29.81, 29.64; MS(ESI):m/z[M+H]+470.13。
2.2 9-(5-氯苯并噻唑-2-甲氧基) 四氢小檗碱(6a)和9-(苯并噻唑-2-甲氧基) 四氢小檗碱(6b)的合成
将9-(5-氯苯并噻唑-2-甲氧基) 小檗碱5a(503 mg, 1 mmol)或9-(苯并噻唑-2-甲氧基) 小檗碱5b(469 mg, 1 mmol)溶解于甲醇(10 mL)中,再向反应体系中加入硼氢化钠(540 mg, 10 mmol)于室温下反应2 h。用薄层色谱监测反应完全,将反应液过滤并将滤液旋干。用石油醚/乙酸乙酯(v/v=10∶1)进行柱层析得到淡黄色固体,产率76%。(6a)1H NMR (400 MHz, Chloroform-d) δ 7.99 (t,J=1.6 Hz, 1H), 7.84 (dd,J=8.5, 1.2 Hz, 1H), 7.38 (dt,J=8.6, 1.6 Hz, 1H), 6.92 (d,J=8.4 Hz, 1H), 6.82 (d,J=8.4 Hz, 1H), 6.72 (s, 1H), 6.58 (s, 1H), 5.91 (s, 2H), 5.51~5.38 (m, 2H), 4.32 (d,J=15.8 Hz,1H),3.85 (s, 3H),3.63~3.48 (m, 2H),3.28~2.76 (m, 4H), 2.69~2.55 (m, 2H);13C NMR (101 MHz, Chloroform-d) δ 171.74, 149.85, 146.05, 128.52, 127.93, 127.87, 125.71, 124.81, 122.97, 122.62, 110.98, 108.53, 105.61, 100.90, 71.62, 59.63, 55.94, 54.21, 51.48, 36.47, 29.81, 29.63; MS(ESI):m/z[M+H]+507.11。 (6b)1H NMR (400 MHz, Chloroform-d) δ 8.02 (dt,J=8.2, 1.0 Hz, 1H), 7.93 (dt,J=7.9, 0.9 Hz, 1H), 7.49 (ddd,J=8.3, 7.2, 1.3 Hz, 1H), 7.40 (ddd,J=8.3, 7.2, 1.2 Hz, 1H), 6.92 (d,J=8.3 Hz, 1H), 6.82 (d,J=8.4 Hz, 1H), 6.72 (s, 1H), 6.58 (s, 1H), 5.91 (s, 2H), 5.54~5.40 (m, 2H), 4.34 (d,J=15.8 Hz, 1H), 3.85 (s, 3H), 3.64~3.49 (m, 2H), 3.29~3.03 (m, 3H), 2.82 (dd,J=15.9, 11.3 Hz, 1H), 2.69~2.53 (m, 2H);13C NMR (101 MHz, Chloroform-d) δ 169.51, 153.02, 149.93, 146.24, 146.03, 143.38, 135.24, 130.79, 128.59, 127.90, 126.17, 125.19, 124.72, 123.15, 121.90, 111.01, 108.53, 105.62, 100.89, 71.73, 59.63, 55.97, 54.23, 51.46, 36.48, 29.81, 29.64; MS(ESI):m/z[M+H]+473.15。
2.3 12-(5-氯苯并噻唑) 四氢小檗红碱(9a)和12-(苯并噻唑) 四氢小檗红碱(9b)的合成
2.3.1 四氢小檗红碱(7) 将小檗红碱2(966 mg, 3 mmol)溶解于甲醇(20 mL)中,再向反应体系中加入硼氢化钠(1 620 mg, 30 mmol)于室温下反应2 h。用薄层色谱监测反应完全,将反应液过滤并将滤液旋干。用石油醚/乙酸乙酯(v/v=10∶1)进行柱层析得到淡粉色固体,产率80%。1H NMR (400 MHz, Chloroform-d) δ 6.74 (t,J=4.2 Hz, 2H), 6.67 (d,J=8.3 Hz, 1H), 6.59 (s, 1H), 5.92 (s, 2H), 4.23 (d,J=15.6 Hz, 1H), 3.87 (s, 3H), 3.61~3.47 (m, 2H), 3.29~3.06 (m, 3H), 2.82 (dd,J=15.9, 11.3 Hz, 1H), 2.72~2.56 (m, 2H)。
2.3.2 12-(甲酰基)四氢小檗红碱(8) 将四氢小檗红碱7(650 mg, 2 mmol)溶解于三氟乙酸(20 mL)中,向反应体系中加入六亚甲基四胺(336 mg, 2.4 mmol)于120 ℃下反应5 h之后,再向其中逐滴加入10%硫酸水溶液(13 mL),并将温度降至90~100 ℃继续反应5 h。反应结束后冷却至室温。用大量饱和碳酸氢钠溶液洗涤,再用乙酸乙酯萃取3次。将萃取得到的有机相收集起来并用无水硫酸镁干燥,之后过滤并将滤液旋干。用二氯甲烷/甲醇(v/v=100∶1)进行柱层析得到红棕色固体,产率21%。1H NMR (400 MHz, DMSO-d6) δ 9.82 (s, 1H), 8.76 (s, 1H), 8.26 (s, 1H), 6.18 (s, 2H), 4.23 (d,J=15.6 Hz, 1H), 3.87 (s, 3H), 3.61~3.47 (m, 2H), 3.29~3.06 (m, 3H),2.82 (dd,J=15.9,11.3 Hz, 1H), 2.72~2.56 (m, 2H)。
2.3.3 12-(5-氯苯并噻唑) 四氢小檗红碱(9a)和12-(苯并噻唑) 四氢小檗红碱(9b) 将12-(甲酰基)四氢小檗红碱8(353 mg, 1 mmol)溶解于N,N-二甲基甲酰胺(10 mL)中,向反应体系中加入2-氨基-4-氯苯硫酚(800 mg, 5 mmol)或者2-氨基苯硫酚(625 mg, 5 mmol),之后再向其中加入水(2 mL)于80 ℃下反应5 h。反应结束后将溶剂旋干,用二氯甲烷/甲醇(v/v=100∶1)进行柱层析得到红棕色固体,产率10%。(9a)1H NMR (400 MHz, DMSO-d6) δ 9.72 (s, 1H), 9.25 (s, 1H), 8.02 (d,J=8.4 Hz, 1H), 7.91 (d,J=2.0 Hz, 1H), 7.47 (s, 1H), 7.39~7.33 (m, 2H), 6.18 (s, 2H), 5.22 (d,J=5.6 Hz, 1H), 4.55 (t,J=6.8 Hz, 4H), 3.81 (s, 3H), 3.18 (d,J=6.3 Hz, 4H);13C NMR (101 MHz, DMSO-d6) δ 171.92, 146.32, 125.71, 121.92, 116.03, 102.32, 56.06, 54.53; MS(ESI):m/z[M+H]+493.09。(9b)1H NMR (400 MHz, DMSO-d6) δ 9.71 (s, 1H), 9.18 (s, 1H), 7.96 (dd,J=8.0, 1.4 Hz, 1H), 7.83 (dt,J=8.1, 0.9 Hz, 1H), 7.47~7.39 (m,2H),7.31~7.24 (m,2H),6.12(s, 2H), 5.23~5.13 (m, 1H), 4.57~4.42 (m, 4H), 3.77 (s, 3H), 3.39~3.45 (m, 2H), 3.12 (t,J=6.1 Hz, 2H);13C NMR (101 MHz, DMSO-d6) δ 167.92, 147.32, 126.77, 121.92, 121.13, 116.03, 102.32, 56.06, 54.53; MS(ESI):m/z[M+H]+459.13。
2.4 13-(5-氯苯并噻唑-2-甲氧基) 小檗碱(11a)和13-(苯并噻唑-2-甲氧基) 小檗碱(11b)的合成
2.4.1 8-乙酰甲基二氢小檗碱(10) 将小檗碱盐酸水合物1(500 mg, 1.4 mmol)溶解于丙酮(10 mL)中,向反应体系中滴加2.3 mL氢氧化钠溶液(5 mol·L-1)于室温下反应1 h。反应完全后,将反应液进行抽滤,并用80%的甲醇水溶液洗涤滤渣2~3次,即得淡黄色固体粗产物,产率85%。MS(ESI):m/z[M+H]+394.16。
2.4.2 13-(5-氯苯并噻唑-2-甲氧基) 小檗碱(11a)和13-(苯并噻唑-2-甲氧基) 小檗碱(11b) 将8-乙酰甲基二氢小檗碱10(393 mg, 1 mmol)和化合物4a(1 085 mg, 5 mmol)或者化合物4b(915 mg, 5 mmol)溶解于乙腈(10 mL)中,再向反应体系中加入碘化钠(750 mg, 5 mmol)和碳酸钾(690 mg, 5 mmol),加热回流反应5 h。反应结束后,过滤除去固体残渣,再将滤液旋干。用二氯甲烷/甲醇(v/v=50∶1)进行柱层析得到黄色固体,产率35%。(11a)1H NMR (400 MHz, DMSO-d6) δ 10.01 (s, 1H), 8.18 (dd,J=8.7, 0.5 Hz, 1H), 8.09 (d,J=9.5 Hz, 1H), 7.93~7.87 (m, 2H), 7.49 (dd,J=8.7, 2.1 Hz, 1H), 7.24 (s, 1H), 7.16 (s, 1H), 6.05 (s, 2H), 5.21 (s, 2H), 4.86 (s, 2H), 4.08 (s, 3H), 3.98 (s, 3H), 3.12 (t,J=5.1 Hz, 2H);13C NMR (101 MHz, DMSO-d6) δ 172.76, 150.92, 150.03, 147.23, 137.55, 134.79, 134.38, 131.84, 126.92, 126.27, 124.54, 122.55, 122.06, 121.60, 120.31, 109.14, 108.69, 102.67, 62.63, 57.45, 49.13, 27.74; MS(ESI):m/z[M+H]+518.10。 (11b)1H NMR (400 MHz, DMSO-d6) δ 10.02 (s, 1H), 8.15 (dt,J=5.3, 3.8 Hz, 1H), 8.09 (d,J=9.4 Hz, 1H), 7.92 (d,J=9.4 Hz, 1H), 7.85~7.77 (m, 1H), 7.45 (dt,J=6.2, 3.6 Hz, 2H), 7.31 (s, 1H), 7.16 (s, 1H), 6.05 (s, 2H), 5.20 (s, 2H), 4.88 (s, 2H), 4.09 (s, 3H), 3.99 (s, 3H), 3.13 (t,J=6.0 Hz, 2H);13C NMR (101 MHz, DMSO-d6) δ 170.12, 152.92, 150.90, 150.00, 147.20, 146.62, 144.77, 137.50, 135.56, 134.75, 133.01, 128.96, 126.98, 126.88, 126.13, 123.06, 123.00, 122.08, 121.63, 120.38, 109.12, 108.68, 102.65, 62.63, 57.43, 35.83, 27.75; MS(ESI):m/z[M+H]+484.14。
2.5 13-(5-氯苯并噻唑-2-甲氧基)四氢小檗碱(12a)和13-(苯并噻唑-2-甲氧基) 四氢小檗碱(12b)的合成
将13-(5-氯苯并噻唑-2-甲氧基) 小檗碱11a(517 mg, 1 mmol)或13-(苯并噻唑-2-甲氧基) 小檗碱11b(483 mg, 1 mmol)溶解于甲醇(10 mL)中,再向反应体系中加入硼氢化钠(540 mg, 10 mmol)于室温下反应2 h。用薄层色谱监测反应完全,将反应液过滤并将滤液旋干。用石油醚/乙酸乙酯(v/v=10∶1)进行柱层析得到淡黄色固体,产率50%。(12a)1H NMR (400 MHz, Chloroform-d) δ 7.93 (d,J=2.1 Hz, 1H), 7.63 (d,J=8.5 Hz, 1H), 7.27 (dd,J=8.5, 2.0 Hz, 1H), 6.72 (s, 1H), 6.61~6.54 (m, 2H), 6.52 (s, 1H), 5.83 (d,J=1.5 Hz, 1H), 5.64 (d,J=1.5 Hz, 1H), 4.23 (d,J=16.1 Hz, 1H), 3.85 (s, 3H), 3.84~3.80 (m, 2H), 3.78 (s, 3H), 3.55 (d,J=16.0 Hz,1H),3.23~3.01 (m,4H),2.68~2.53 (m, 2H);13C NMR (101 MHz, Chloroform-d) δ 174.03, 154.31, 150.76, 146.38, 146.09, 144.96, 133.85, 131.63, 131.40, 129.84, 128.67, 128.38, 124.85, 124.49, 122.42, 122.02, 110.42, 108.35, 106.18, 100.85, 63.26, 60.18, 55.76, 54.39, 51.15, 44.97, 37.51, 30.27; MS(ESI):m/z[M+H]+521.13. (12b)1H NMR (400 MHz, Chloroform-d) δ 7.95 (dt,J=8.0, 0.9 Hz, 1H), 7.80~7.66 (m, 1H), 7.41 (ddd,J=8.3, 7.2, 1.3 Hz, 1H), 7.29 (ddd,J=8.2, 7.2, 1.2 Hz, 1H), 6.74 (s, 1H), 6.58 (d,J=1.5 Hz, 2H), 6.53 (s, 1H), 5.81 (d,J=1.5 Hz, 1H), 5.58 (d,J=1.5 Hz, 1H), 4.24 (d,J=16.0 Hz, 1H), 3.87 (dd,J=6.4, 2.8 Hz, 1H), 3.85 (s, 3H), 3.82 (d,J=2.9 Hz, 1H), 3.78 (s, 3H), 3.56 (d,J=16.0 Hz, 1H), 3.24~3.04 (m, 4H), 2.66~2.53 (m, 2H);13C NMR (101 MHz, Chloroform-d) δ 171.98, 153.46, 150.70, 146.38, 146.06, 144.93, 135.59, 131.58, 129.83, 128.63, 128.46, 125.65, 124.62, 124.40, 122.54, 121.34, 110.42, 108.33, 106.24, 100.79, 63.34, 60.18, 55.76, 54.44, 51.20, 45.04, 37.41, 29.91; MS(ESI):m/z[M+H]+487.16。
3 体外抗菌活性实验
使用液体稀释法测定小檗碱及其衍生物的最低抑菌浓度(Minimum Inhibitory Concentration,MIC),具体操作如下:在96孔细胞培养板中,用液体培养基等倍稀释小檗碱及其衍生物的药液(浓度为32、16、8和4 μg·mL-1),每孔加100 μL稀释药液和10 μL供试菌液,于37 ℃培养24 h后用MTT法检测在600 nm下各孔的吸光度值。同步设定培养基对照孔、药液对照孔、菌液对照孔,在培养基对照孔无菌生长和菌液对照孔有菌生长的情况下,分析判断实验结果,以OD值突然升高前的临界值为药物最低抑菌浓度。
4 分子对接
以FtsZ蛋白(PDB: 4DXD)为模板,采用Ledock软件分析化合物与FtsZ蛋白的结合模式,使用ChemBio3D Ultra 14.0软件处理小檗碱及其衍生物的3D结构,使其MM2能量最小化。采用LePro处理FtsZ蛋白结构使其适用于分子对接。根据文献报道推测可能性最大的结合位点,使用LeDock软件将化合物与FtsZ蛋白进行柔性对接,打分最高的结合构象以PyMOL软件和薛定谔软件处理后展示。
5 讨论
依据上述研究方向共合成出10个小檗碱衍生物,大体上可分为3条路线(见图3)。目标化合物5a、5b、6a、6b的合成方法可归纳为一条路线:首先将小檗碱(1)在真空高温条件下脱去9位的甲基生成小檗红碱(2)[13-14]。化合物3与氯乙酰氯成环得到化合物4,4再与小檗红碱(2)发生亲核取代反应即得目标化合物5[15-16]。将化合物5用硼氢化钠进行还原,即得目标化合物6[17-19]。目标产物9a和9b的合成:首先也是将小檗碱(1)在真空高温条件下转化成小檗红碱(2),再利用硼氢化钠对其还原得到四氢小檗红碱(7)。之后将化合物7与六亚甲基四胺进行一步甲酰化反应生成12-(甲酰基)四氢小檗红碱(8)[20-21],化合物8再与2-氨基-4-氯苯硫酚或2-氨基苯硫酚反应即可得到目标化合物9a或9b[22]。目标化合物11a、11b、12a、12b的合成:将小檗碱与丙酮反应生成中间体10,再将其与化合物4a或4b反应即可得到目标化合物11a或11b[23]。将化合物11用硼氢化钠进行还原,即得目标化合物12。通过实验验证这3条路线都是切实可行的,优点是操作简单,原料易得;缺点是产率较低。尤其是目标化合物9的产率只有10%,后期还需要进一步优化合成方法。
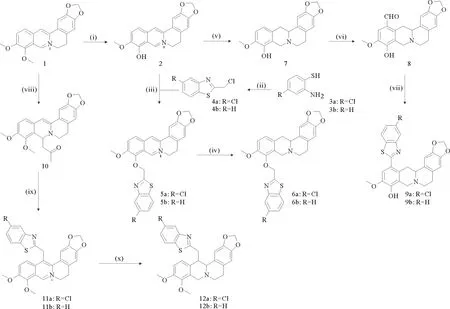
(反应试剂和条件:(i)190 ℃,真空,1 h;(ii)氯乙酰氯,三乙胺,甲苯;(iii)碘化钠,碳酸钾,乙腈,加热回流;(iv)硼氢化钠,甲醇,室温,21 h;(v)硼氢化钠,甲醇,室温,21 h;(vi)Ι.六亚甲基四胺/三氟醋酸,120 ℃;Ⅱ.百分之十的硫酸水溶液,90~100 ℃;(vii)3a/3b,N, N-二甲基甲酰胺/水,80 ℃;(viii)氢氧化钠水溶液(5 mol·L-1),丙酮,室温,11 h;(ix)4a/4b,碘化钠,碳酸钾,乙腈,加热回流;(x)硼氢化钠,甲醇,室温,21 h。Reagents and conditions: (i) 190 ℃, in vacuo, 1 h; (ii) ClCOCH2Cl, Et3N, toluene; (iii) NaI, K2CO3, CH3CN, reflux; (iv) NaBH4, MeOH, r.t., 2 h; (v) NaBH4, MeOH, r.t., 2 h; (vi) Ι. HMTA/TFA, 120 ℃; Ⅱ. 10% H2SO4, 90~100 ℃; (vii) 3a/3b, DMF/H2O, 80 ℃; (viii) 5 N NaOH, acetone, r.t., 1 h; (ix) 4a/4b, NaI, K2CO3, CH3CN, reflux; (x) NaBH4, MeOH, r.t., 2 h.)
对合成出的10个小檗碱衍生物进行体外抗菌活性实验,结果表明化合物5a、5b、9a、11a、11b对两种革兰氏阳性菌(S.aureusATCC 6358和MRSA ATCC 43300)的抗菌效果均优于小檗碱(见表1),其中化合物11a抑菌效果最佳(抗菌活性相较于小檗碱提高了16倍),可能是因为小檗碱13-位引入5-氯苯并噻唑片段后更有利于其和FtsZ蛋白相结合。为了进一步探究化合物11a抗菌活性提高的原因,用Ledock软件对其进行分子对接(见图4A、4B),对接结果表明小檗碱衍生物结合于FtsZ蛋白N端GTP酶区域的疏水口袋中。相较于小檗碱而言,化合物11a不仅能与Thr133、Asn166氨基酸残基形成氢键相互作用,而且其分子构型更适合FtsZ蛋白N端结合域的疏水空腔,这可能是其抗菌活性提高的原因。
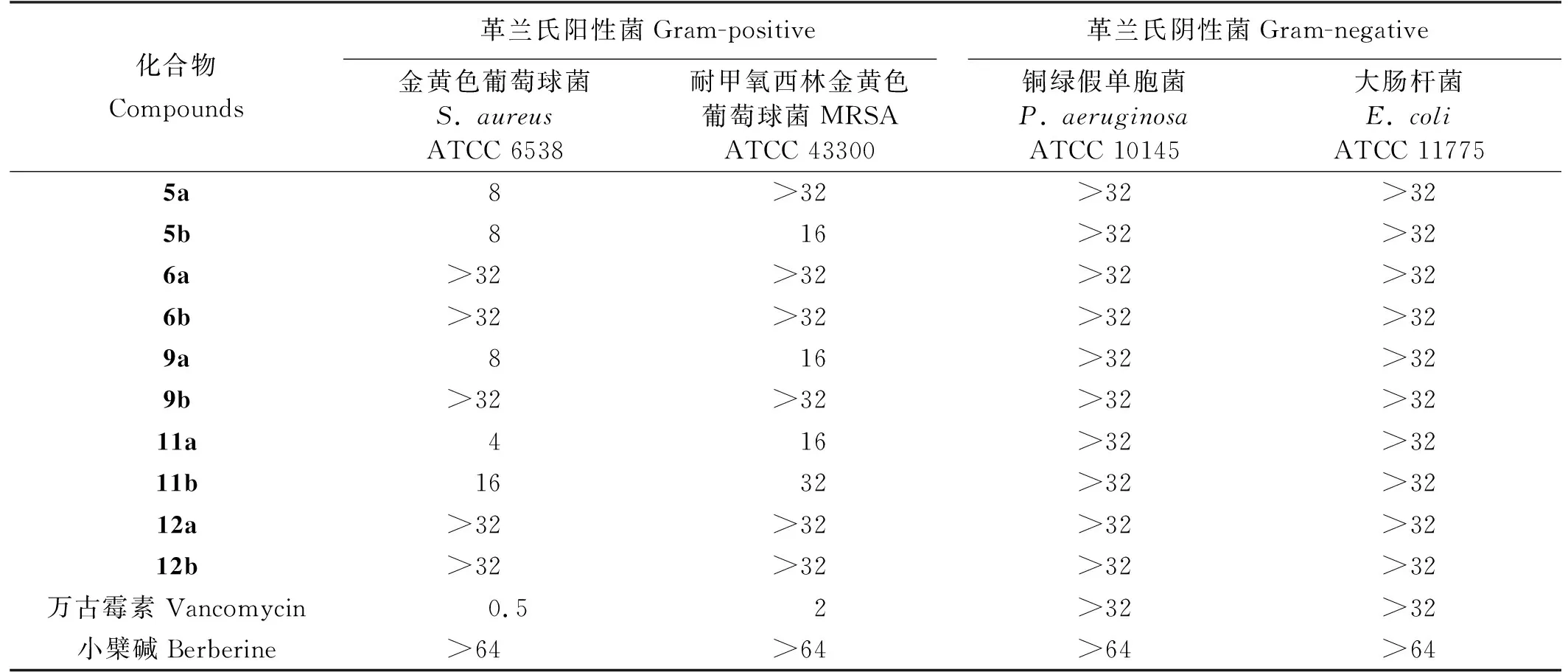
表1 小檗碱衍生物体外抗菌活性实验(最小抑菌浓度:微克/毫升)

图4 分子对接(PDB: 4DXD)
6 结语
本文旨在将FtsZ抑制剂PC190723衍生物的苯并噻唑类活性片段与小檗碱相结合以提高小檗碱的抗菌活性。依据此研究方向共合成出9-位、12-位、13-位不同位置取代的10个小檗碱衍生物并对其进行了体外抗菌活性测试。抗菌活性数据显示化合物5a、5b、9a、11a、11b对两种革兰氏阳性菌(S.aureusATCC 6358和MRSA ATCC 43300)的抗菌效果均优于小檗碱(抗菌活性分别提高了8、8、8、16和4倍),初步说明小檗碱衍生物具有成为靶向FtsZ抗多药耐药的新型抗菌药物的潜力。
猜你喜欢
云南化工(2021年7期)2021-12-21
现代临床医学(2021年4期)2021-07-31
西南农业学报(2020年8期)2020-12-10
吉林农业(2019年6期)2019-06-11
中成药(2018年12期)2018-12-29
中成药(2018年3期)2018-05-07
中成药(2017年10期)2017-11-16
中成药(2017年4期)2017-05-17
郑州大学学报(工学版)(2015年1期)2015-03-24
食品科学(2013年24期)2013-03-11
