
一例1q25.1-q31.1杂合缺失患儿临床及遗传学分析并文献复习
2024-02-20 04:57陆相朋
郑州大学学报(医学版) 2024年1期
王 宇,郑 宏,陆相朋
1)河南中医药大学儿科医学院 郑州 450099 2)河南中医药大学第一附属医院儿科医院 郑州 450099
染色体1q缺失相对罕见,其常见的临床表现包括产前和产后生长缺陷、发育迟缓、智力残疾、小头畸形、短头畸形和骨骼异常[1]。Taysi等[2]根据缺失位置将1q缺失分为近端缺失(1q21-22→q25)、中间缺失(1q24-25→q32)以及末端缺失(1q42-43→末端)3种类型。本文报道1例患儿,以宫内发育迟缓、生长迟缓、精神运动迟缓、喂养困难、小头畸形、小颌畸形、双眼内斜视、肌张力低下为主要临床特点,经全基因组测序分析为1q中间缺失。
1 病例资料
1.1 一般情况患儿,女,1岁8个月,因“出生后生长发育迟缓至今”入院。患儿出生后混合喂养,自新生儿期身高、体重低于正常同龄儿,3个月竖头稍差,4个月会翻身,6个月独坐不稳,6个月龄后身高、体重未见明显增长,1岁不会独走,不会说话,生长发育较同龄儿落后,家长未予重视;1岁5个月于当地医院行头颅MRI示双侧大脑半球多发脑白质脱髓鞘改变,行综合康复治疗2个月进步缓慢,为求进一步诊治,遂来河南中医药大学第一附属医院就诊,门诊以“弥漫性发育障碍”为诊断收入儿科。入院症见:患儿可扶走,不能独走,可发简单音节,会听指令。患儿母亲怀孕2次生产2次,孕32周彩超提示胎儿偏小,足月剖宫产。出生时身长48 cm(<-1SD),体重2.6 kg (<-2SD ),出生后4 d黄疸加重,嗜睡,喂养困难,进一步检查发现脐部感染,行细菌培养未见明显细菌生长(具体不详),经治疗后好转出院。患儿父母体健,一姐体健,否认家族遗传病史。
入院体格检查:身高72 cm(<-3SD),体重8 kg(<-3SD),头围42.8 cm(<-3SD),神志清,精神可,营养不良貌。皮肤、毛发黄。表观异常:舟状头,前额凸,面中线凹陷,山峰眉,双眼内斜视,眼裂宽,长睑裂,下眼睑轻度外翻,外眼角上斜(轻度),鼻根低平,圆头鼻,鼻翼宽,下颌短小,齿列不齐。钟状胸,乳距正常,心、肺、腹部查体未见异常,外阴无异常。追视、追听可,可逗笑,能笑出声;有意识发“yiya”音,与人眼神交流,呼之反应,双手有主动抓物意识,灵活性可,竖头可,会翻身,可坐稳。仰卧位:头部居中,四肢对称;俯卧位:肘支撑,可抬头90°;坐位:可坐稳;立位:双下肢可短暂支持体重,外展外旋,足外翻,膝过伸。肌力、肌张力检查:四肢肌力4级,肌张力低,下肢双侧股角160°,腘窝角160°,踝关节被动活动无抵抗,双足背屈角快角90°~100°,慢角70°。双侧膝腱反射可引出;病理反射:双侧巴宾斯基征(-);踝阵挛(-),脑膜刺激征(-)。
实验室检查:血尿常规、甲状腺功能、肝肾功能、电解质、心肌酶、葡萄糖、生长激素、胰岛素等生化检查未见异常。血氨基酸及肉碱谱分析、尿有机酸分析未见异常。心脏、肝、胆、胰、脾、肾彩超检查未见异常。头颅MRI示双侧大脑半球多发脑白质脱髓鞘改变。
Gesell发育量表评估(1岁7个月龄):适应能力37.3分,相当于6.8个月;粗大运动37.3分,相当于6.8个月;精细动作37.9分,相当于6.9个月;言语能力32.4分,相当于5.9个月;社交能力48.8分,相当于8.9个月;综合发育商38.7分,整体发育迟滞。
1.2 基因检测为进一步明确诊断,与患儿父母沟通并获得知情同意后,经河南中医药大学第一附属医院医学伦理委员会批准(伦理审批号:AF/SL-02/03.1),采集先证者外周血2 mL提取DNA,送北京全谱医学检验实验室,行线粒体DNA高敏感性测序分析及全基因组测序。最后由临床医生、遗传学专家共同做出评判。
线粒体DNA高敏感性测序分析未见异常。
全基因组测序结果:46,XX,del(1q25.1-q31.1)(174 297 018~188 975 050)×1。该区段基因拷贝数变异在普通人人类基因变异数据库中未见报道,Decipher患者库中多个可能致病性杂合缺失变异与该基因拷贝数变异区间部分重叠。因本次检测缺少家系成员(尤其是其父母)携带信息,故无法判断遗传共分离,根据《美国医学遗传学与基因组学学会指南》综合分析,评级为“可能致病”。
最终诊断为1q中间缺失综合征。
1.3 文献复习以“1号染色体缺失”“1q缺失”“1q deletion”为关键词在中国知网、万方医学网、PubMed数据库中检索筛选相关文献,1974至2022年收集到存在1q25.1-q31.1缺失患儿35例,其中中国患儿4例。对35例已报道及本研究病例的基因组缺失区域及大小、临床表型的总结见表1。1q中间缺失的常见临床特征包括宫内生长迟缓(24/32)、生长迟缓(23/30)、精神运动迟缓(23/28)、小头畸形(17/30)、第五指弯曲(20/33)、生殖器异常(18/32)、唇/腭异常(16/33)、短指/趾(17/31)、心脏异常(13/31)、小手和小脚(12/28)、小颌畸形(8/30)、肾脏异常(8/30),另外甲状腺功能减退、生长激素缺乏、斜视、短鼻和球根鼻、少牙、耳郭畸形、颅脑发育畸形也有报道。
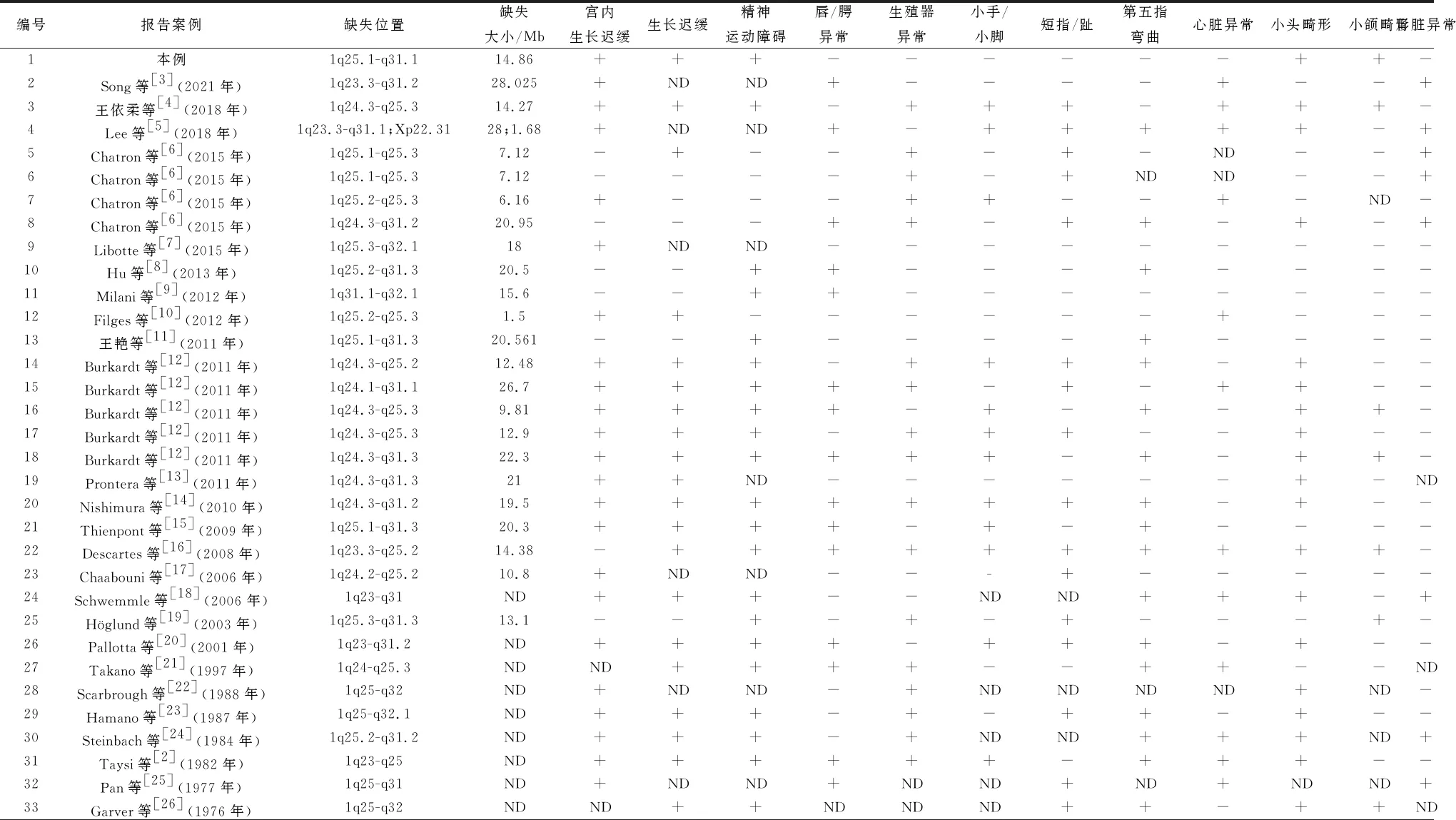
表1 1q25.1-q31.1缺失患儿临床表型比较
2 讨论
染色体异常与儿童生长迟缓关系密切[29]。本文分析结果显示,尽管临床表现与缺失区域相关,但临床特征和1q缺失片段大小不存在平行性,携带不同程度缺失的患儿可表现出相似的表型[7]。另外,许多临床体征是非特异性的,与在1q近端缺失或1q远端缺失患儿中观察到的体征存在一定的重叠,因此很难将某些性状与缺失的1q片段一一对应[19]。几乎所有1q24-25→q32缺失的患儿都有严重的产前和(或)产后生长迟缓。该区域含有LHX4和CENPL等重要生长基因,其缺失往往会导致生长迟缓及发育缺陷。LHX4基因对垂体和神经发育具有重要调控意义,其杂合突变可导致垂体发育不全、小脑异常、生长激素缺乏,进而导致生长迟缓[30]。CENPL基因对正常的动粒功能和有丝分裂进程尤为重要,该基因的缺失会导致生长缺陷[31]。值得注意的是,仅LHX4基因缺失也会出现表型正常的情况,而所有CENPL杂合缺失的报道都有生长迟缓的表现,因此CENPL杂合缺失表现为对生长迟缓的完全外显,而LHX4缺失则表现为不完全外显[8]。本例患儿仅存在LHX4基因缺失,考虑为患儿生长迟缓的重要原因。
Hu等[8]通过整合11例1q25-q32缺失且智力障碍的患儿,提出1q25.2 中的3.1 Mb为关键区域,并猜测杂合性ASTN1缺失是1q24-25→q32缺失患儿智力障碍的一个原因。ASTN1编码一种神经元黏附分子,该分子对于神经胶质引导下发育中大脑皮质区域神经元的迁移至关重要[32],ASTN1突变可能是导致包括脑裂、脑干增粗、皮质异常增厚、侧脑室扩大、胼胝体/小脑半球发育不良一系列皮质畸形的原因[33]。而Burkardt等[12]认为DNM3基因是智力残疾的相关基因,DNM3基因编码Dynain-3蛋白,该蛋白在成人的大脑和脊髓中高表达,可与突触后Homer蛋白结合,促进谷氨酰胺能神经元成熟并增加突触强度[34]。Trinh等[35]的研究表明,DNM3是帕金森综合征发病年龄的遗传修饰因子,而后多项研究[36-38]却得出相反的结果。因此DNM3基因对智力发育的影响亟待深入的研究。本例患儿存在智力发育迟缓,考虑与位于1q25.2的ASTN1基因的缺失有关。
此外,1q24-25→q32区域内ASPM基因突变可引起原发性小头畸形5型,其临床特征是胎儿期脑发育异常,导致脑容量减少,头围减小和智力发育不全,由胚胎神经发育过程中产生的大脑皮层神经元数量减少所致[39]。小头畸形病因多而复杂,本例患儿并未缺失ASPM,而缺失的GLUL致病基因与神经发育相关,其在全身表达,从神经元突触中清除L-谷氨酸,是中枢神经系统中的主要神经递质[40]。
第五指弯曲等异常骨骼表现可能与PRG4杂合缺失相关。PRG4基因编码存在于滑液和关节软骨表面的润滑糖蛋白,该蛋白可保护软骨表面[41],润滑糖蛋白突变体Prg4-/-小鼠的关节会出现形态学改变[42]。PRG4基因缺失或突变与弓背关节病-髋内翻-心包炎综合征的发生有关,多表现为先天性指屈曲或足趾屈曲[43]。本例患儿存在PRG4基因缺失,但体格检查未发现第五指弯曲,可能与检查时机或个体差异有关。
综上所述,染色体1q缺失的临床特征复杂程度与缺失片段大小并不平行。1q中间缺失(1q24-25→q32)的表型相关候选基因有LHX4、CENPL、ASTN1、DNM3、ASPM、GLUL、PRG4等,推测此类患儿表型与缺失片段基因型有关,但还需更多证据来支持。
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
现代仪器与医疗(2022年1期)2022-04-19
种子(2021年3期)2021-04-12
现代园艺(2017年21期)2018-01-03
中国医药指南(2017年3期)2017-11-13
外语教学理论与实践(2016年1期)2016-06-11
中国康复理论与实践(2015年10期)2015-12-24
医学研究杂志(2015年5期)2015-06-10
现代检验医学杂志(2015年5期)2015-02-06
华东理工大学学报(自然科学版)(2014年1期)2014-02-27
