
基于晶格匹配Pt-Co-ZnO三元界面催化剂设计及其催化CO2加氢性能
2024-02-21 03:13万雅琴张煜华李金林
石油炼制与化工 2024年2期
万雅琴,张煜华,李金林,王 立
(中南民族大学化学与材料科学学院,催化转化与能源材料化学教育部重点实验室,催化材料科学湖北省重点实验室,武汉 430074)
化石燃料驱动着人类社会的进步与发展,同时也伴随着二氧化碳(CO2)排放的日益增长[1-3]。2022年全球CO2浓度达到了417.2 μg/g,创历史新高。将CO2进行捕获并加以利用,不仅能减少CO2排放量,还能将CO2转化为高附加值的化学品。CO2加氢催化转化是CO2捕获利用最重要的技术之一,能同时解决环境问题、缓解能源危机[4-6]。
众所周知,CO2分子热稳定性高,难以活化;同时CO2催化加氢反应机理十分复杂,副反应众多,导致了反应的选择性难以被精确调控[7-8]。这些因素均对催化剂结构的设计提出了更高的要求。三元界面催化剂因其各功能组分间具有强相互作用和电子转移特性,在催化CO2加氢反应上表现出巨大的应用潜力[9-10]。Pinheiro等[11]证实在Pd-In2O3-ZrO2三元催化剂中,三组分的微界面环境对其催化CO2加氢制甲醇至关重要。Wang Yuhao等[12]也结合实验和密度泛函理论(DFT)计算,证实了Cu-ZnO-ZrO2催化剂中三组分微界面环境对于CO2活化和产物醇的生成起重要作用。三元界面催化剂性能优异的原因在于其高密度的界面位点能促进CO2活化,改变关键反应途径。然而,精准构建三元界面仍然是该类催化剂设计制备的挑战之一[13]。在采用传统浸渍法或沉积沉淀法制备多组分催化剂的过程中,一般需要高温焙烧过程将金属盐前体转化为氧化物颗粒。高温过程可能造成组分分离或形成氧化物合金,不利于界面的精确构建。
钴基催化剂因具有较强的CO2活化能力和C—C偶联能力被寄予厚望[14-17]。作为CO2加氢催化剂,通过调控物相结构能够定向得到甲烷(CH4)或一氧化碳(CO)等不同产物。ZnO因具有较强的解离H2分子的能力而常常被用作CO2加氢多组分催化剂中的结构助剂[18]。金属Pt不仅能通过氢溢流作用促进过渡金属还原,自身也拥有较强的CO2活化能力,被广泛用于CO2催化加氢反应[19-25]。
基于三元微界面环境对催化剂性能的重要作用和CoO、ZnO、金属Pt对不同反应的催化特性,本研究基于晶格匹配的策略设计Pt-ZnO@CoO核壳结构三元催化剂[26],并对其进行还原预处理,使Pt颗粒分散在Co颗粒周围,且两种还原金属颗粒同时负载在ZnO纳米棒表面,形成三元微界面环境,进而考察其催化CO2加氢反应的性能,以期研制一种高性能的CO2加氢催化剂,为将CO2转化成高附加值的化学品提供技术支撑。
1 实 验
1.1 原料和试剂
六亚甲基四胺(C6H12N4)、二水乙酸锌(C4H6O4Zn·2H2O)、无水乙醇(C2H5OH),均为分析纯,国药集团化学试剂有限公司产品;乙酰丙酮钴(Ⅱ)(C10H14O4Co)、乙酰丙酮钴(Ⅲ)(C15H21CoO6)、苄胺(C7H9N)、氯铂酸溶液(H2PtCl6,质量分数为8%),均为分析纯,阿拉丁公司产品。
1.2 ZnO纳米棒的制备
采用溶液沉淀法制备ZnO纳米棒[27]。具体步骤如下:将2.90 g六亚甲基四胺和3.00 g二水乙酸锌加入1 000 mL圆底烧瓶中,加入去离子水800 mL;搅拌3 min,再超声混合3 min,反复3次,保证混合均匀;然后放入90 ℃油浴中回流反应12 h,再静置12 h;最后用无水乙醇离心洗涤3次,200 ℃下干燥2 h,得到ZnO纳米棒。
1.3 hcp-CoO的制备
采用热分解法制备hcp-CoO。具体步骤如下:将1.60 g乙酰丙酮钴(Ⅲ)和96.00 g苄胺加入250 mL圆底烧瓶中,先在135 ℃油浴中预热5 min,然后转入190 ℃油浴(闪热)反应1.5 h,冷却后用无水乙醇离心洗涤3次,并在60 ℃下干燥2 h后得到hcp-CoO。
1.4 ZnO@CoO的制备
基于晶格匹配,采用热分解法将Co物种定向沉积到ZnO纳米棒表面,合成了ZnO@CoO纳米包覆材料,其具体制备步骤如下:将1.70 g上述制备的六方棒状ZnO纳米棒和0.30 g的乙酰丙酮钴(Ⅱ)加入到250 mL圆底烧瓶中,加入100.00 g苄胺;搅拌3 min,再超声混合3 min,反复3次,保证混合均匀。然后放入185 ℃油浴中回流反应1.5 h,反应完毕后冷却,用无水乙醇离心洗涤3次,100 ℃下干燥2 h,得到ZnO@CoO催化剂。
1.5 Pt/ZnO和Pt-ZnO@CoO的制备
贵金属Pt的引入采用浸渍法,具体步骤如下:先进行水饱和试验,确定1.00 g ZnO或ZnO@CoO的饱和吸水量,然后按理论配比量取氯铂酸溶液(H2PtCl6,质量分数为8%),并根据水饱和试验结果配制成一定体积的水溶液;将配制的氯铂酸水溶液逐滴浸渍在ZnO或ZnO@CoO上,浸渍完成后旋蒸,100 ℃下干燥过夜,得到Pt/ZnO或Pt-ZnO@CoO催化剂。
1.6 催化剂表征
催化剂形貌表征通过日本日立公司生产的SU8010型扫描电子显微镜(SEM)进行;催化剂尺寸、形貌及元素分布利用美国Thermo Fisher公司制造的Talos F200X型扫描透射电子显微镜(STEM)表征。
利用德国Bruker公司生产的Advance D8型X-射线粉末衍射仪(XRD)对催化剂进行物相分析:Cu Kα、Vantec-1探测器、在40 kV和40 mA下进行测试,扫描范围10°~80°,扫描步长为2(°)/min。
催化剂元素含量采用美国Flexar公司生产的NEXLON 300X型电感耦合等离子质谱仪(ICP-MS)测得。测试时,将少量焙烧后的样品溶解在浓硝酸中,并用超纯水稀释至不同浓度梯度后进行测试。
利用美国Zeton Altmira公司生产的AMI-200型多功能表征仪对催化剂进行H2程序升温还原(H2-TPR)和CO2程序升温脱附(CO2-TPD)测试,表征其还原性质和CO2吸附能力。首先称取约50 mg样品置于U型样品管中部,底部和上部放入石英棉,然后将样品管装进仪器加热炉中:①H2-TPR测试前先通入Ar(30 mL/min),并以5 ℃/min的速率升温至150 ℃,保持30 min以除去样品中的水;待样品冷却至50 ℃后,以流量30 mL/min通入5% H2(φ)-95%Ar(φ)混合气吹扫30 min,以走平基线;然后以10 ℃/min的速率升温至700 ℃并保持30 min。②CO2-TPD测试前先通入He(25 mL/min),并以10 ℃/min的速率升温至150 ℃,保持1 h以除去催化剂中的水和氧,然后降温至50 ℃,通入H2对样品进行还原,并以10 ℃/min的速率升温至250 ℃,保持3 h;完成后通入He进行吹扫降温至100 ℃,以25 mL/min的流量通入10% CO2(φ)-90% He(φ)混合气进行吸附,保持60 min;吸附完成后通入He(25 mL/min)吹扫30 min,以吹平基线,然后以10 ℃/min的速率从100 ℃升温至600 ℃并保持45 min,进行脱附。测试过程通过热导池检测器记录信号,得到相应的脱附曲线。
1.7 CO2加氢反应性能评价
催化剂的CO2加氢反应性能评价在自建的固定床反应器上进行。该反应器的反应管长53 cm,内径8 mm。性能测试开始前,先在常压、250 ℃、空速为6 L/(gcat·h)的H2气氛中对催化剂预处理3 h,再通入反应混合气22.5%CO2(φ)-67.5%H2(φ)-10.0%N2(φ),使反应器压力到达2.0 MPa,并以1 ℃/min的速率升温至目标反应温度,进行反应。反应过程中的气相产物通过Agilent公司生产的GC 3000型气相色谱在线分析,液相产物经冷阱冷凝后通过Agilent公司生产的GC 4890D气相色谱离线分析。
根据产物气相色谱分析结果,计算CO2转化率(XCO2)、CH4选择性(SCH4)、CO选择性(SCO)、Cn选择性(SCn)、C2+选择性(SC2+)、甲醇选择性(SCH3OH),其计算见式(1)~式(6)。

(1)

(2)

(3)

(4)
SC2+=SC2+SC3+SC4+SC5+SC6+SC7+SC8
(5)
SCH3OH=100%-SCO-SCH4-SC2+
(6)
2 结果与讨论
2.1 形貌表征和XRD表征
图1为制得ZnO纳米棒的SEM照片。从图1可以看到,所制的ZnO形貌为比较均一的纳米棒,长度为2~5 μm,直径为200~400 nm,截面呈现规整的六边形,为典型的六方晶型ZnO形貌,其6个侧面为六方晶型的(100)晶面,两个底面为六方晶型的(002)晶面[27]。

图1 制得ZnO纳米棒的SEM照片
基于晶格匹配策略,采用热分解法将Co物种定向沉积到ZnO纳米棒表面,合成了ZnO@CoO纳米包覆材料以及相应的改性催化剂。图2为前面所制催化剂的XRD图谱。由图2可以看到:XRD图谱中没有出现Pt的特征衍射峰,说明Pt在催化剂表面高度分散;所制催化剂都具有六方晶型ZnO的特征衍射峰,而且hcp-CoO与ZnO的特征衍射峰几乎完全重叠,表明两者具有非常相似的晶相结构。两者的晶格失配度可以通过式(7)计算:
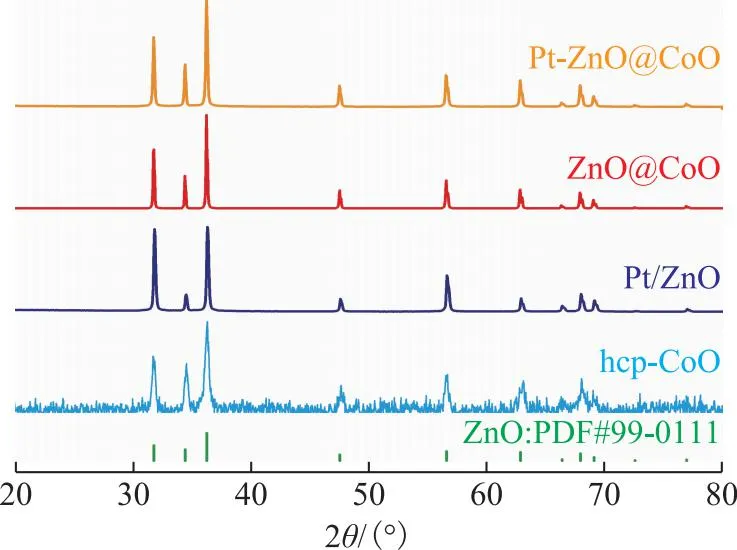
图2 所制催化剂的XRD图谱
f=(αs-αg)/αs
(7)
式中:f为晶格失配度;αs和αg分别为晶种与外延层材料的晶胞参数。
通过晶胞参数计算可知,二者的晶格失配度为0.2%,故二者的晶格匹配度为99.8%,见表1。

表1 以XRD 数据计算的ZnO和hcp-CoO的晶胞参数
ICP结果显示,ZnO@CoO和Pt-ZnO@CoO催化剂Co元素的质量分数为2.7%。进而,通过STEM及能量散色X射线光谱(EDX)表征ZnO@CoO,Pt/ZnO,Pt-ZnO@CoO催化剂及其各元素分布,结果如图3所示。由图3(b)可以看到,CoO均匀地包覆在ZnO纳米棒外围,厚度为8 nm左右。由于hcp-CoO与ZnO之间具有超高的晶格匹配度,hcp-CoO在ZnO表面形成了连续的包覆层,这对于形成三元界面至关重要。对于Pt-ZnO@CoO催化剂,由图3(f)可以看到,Pt均匀地分布在材料表面,同时Pt的沉积并没有破坏CoO层。同样,在Pt/ZnO催化剂中,Pt也均匀地分散在ZnO载体表面上,见图3(j)。STEM结果与XRD结果一致,说明Pt在催化剂表面高度分散。

图3 ZnO@CoO,Pt/ZnO,Pt-ZnO@CoO催化剂的STEM照片和EDX元素分布
2.2 H2-TPR表征
利用H2-TPR表征了不同催化剂的还原性质,结果如图4所示,对于ZnO@CoO催化剂,280 ℃的还原峰归属于表面四氧化三钴(Co3O4)的还原,370 ℃的还原峰归属于CoO的还原,450 ℃左右的还原峰归属于与ZnO匹配的CoO的还原。对于Pt-ZnO@CoO催化剂,190 ℃的还原峰归属于Pt前躯体盐的分解,210~400 ℃的还原峰归属于CoO的还原[19,28],该还原峰对应的温度明显低于ZnO@CoO催化剂中CoO还原温度。Pt具有较强的氢解离能力,解离的氢可以通过氢溢流效应转移到CoO表面,促进其还原。对于Co基催化剂而言,Co物种的状态对于催化剂的活性和选择性影响很大。当催化剂还原度较高时,反应过程中Co物种主要以金属Co的形式存在,催化剂活性较高,主要产物为CH4。当催化剂还原度较低时,CoOx为主要的活性相,这时候催化剂活性较低,CO2加氢主要产物为CO[18]。H2-TPR结果说明Pt促进了CoO的还原,这对于提升催化剂性能至关重要。
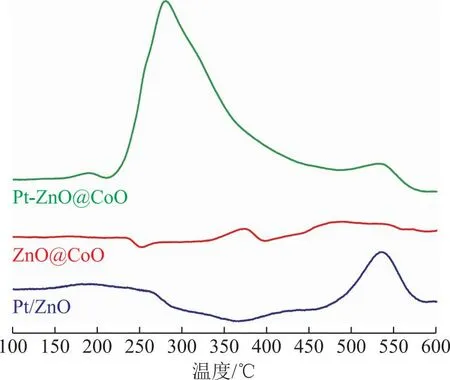
图4 催化剂的H2-TPR表征结果
2.3 CO2加氢反应性能评价
在固定床反应器上考察上述3种催化剂对CO2加氢反应的催化性能,结果如图5所示;不同反应温度下Pt-ZnO@CoO催化剂的CO2转化率及产物选择性见表2。从图5(a)可以看到,ZnO@CoO与Pt/ZnO催化剂在300 ℃时CO2转化率几乎为零,说明这两种催化剂对CO2加氢反应没有催化活性。而Pt-ZnO@CoO催化剂在160 ℃就开始表现出催化活性,此时CO2转化率为0.4%,产物醇的选择性为95.2%,唯一气相产物CH4的选择性为4.8%;随着反应温度升高,CO2转化率逐渐升高,但产物醇的选择性逐渐下降;当反应温度为300 ℃时,CO2转化率为20.1%,CH4的选择性为70.3%,CO的选择性为2.8%,醇的选择性仍有26.3%。此外,Pt-ZnO@CoO催化剂在55 h内 CO2转化率基本不变,说明该催化剂具有良好的稳定性,见图5(b)。
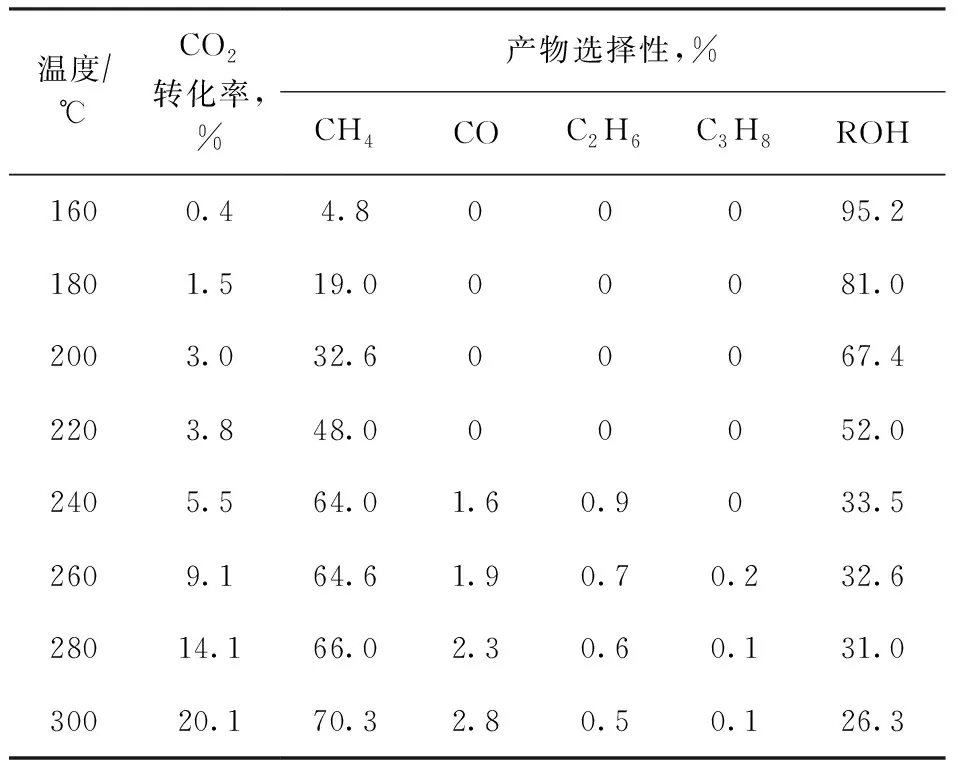
表2 不同反应温度下Pt-ZnO@CoO催化剂的CO2转化率及产物选择性
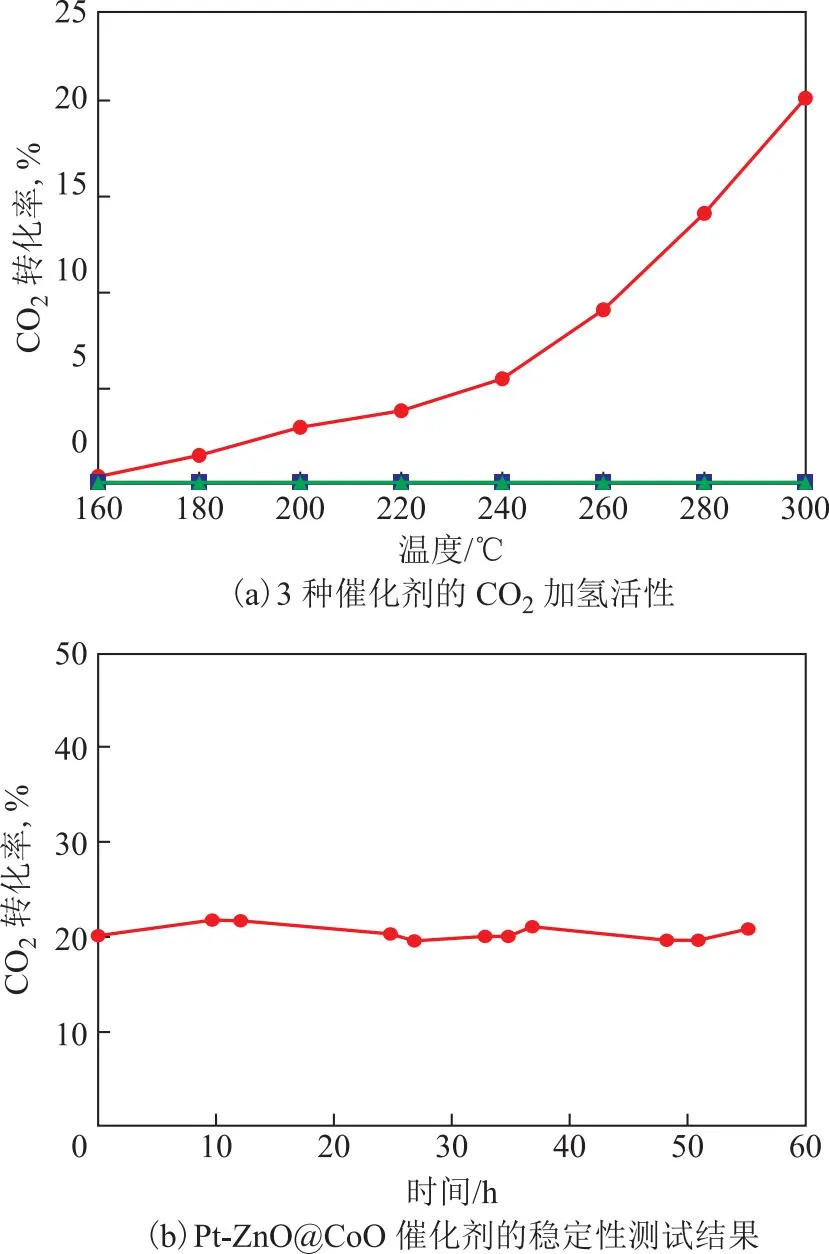
图5 催化剂催化CO2加氢反应性能评价结果●—Pt-ZnO@CoO; ■—ZnO@CoO; ▲—Pt/ZnO
除了逆水煤气变换反应(RWGS),大多数 CO2加氢路线都是放热反应,热力学上低温对反应有利;但是由于CO2具有较强的热稳定性和化学惰性,需要足够高的温度将其活化。经过计算发现,烷烃(如甲烷等)是热力学上有利的产物,而醇的生成需要由动力学主导,因此探索醇选择性高的催化剂具有重要的意义[1]。
2.4 还原后Pt-ZnO@CoO催化剂的形貌表征
为了阐明上述催化剂催化性能存在差异的原因,对还原后的Pt-ZnO@CoO催化剂进行了SEM表征,结果见图6。由图6可以看到,在250 ℃下H2还原3 h之后,催化剂样品的六方棒状形貌保存完好,样品表面出现明显的颗粒,其分散相对均匀,颗粒粒径为20~40 nm,说明Co纳米颗粒均匀分散在ZnO纳米棒表面,形成了纳米复合结构。

图6 还原后Pt-ZnO@CoO催化剂的SEM照片
图7为该还原后催化剂的STEM和高分辨透射电子显微镜(HRTEM)照片及对应的EDX元素分布。图7进一步印证了还原后的催化剂纳米棒上出现了金属Co颗粒。

图7 还原后Pt-ZnO@CoO催化剂的电子显微镜照片和EDX元素分布
2.5 反应后Pt-ZnO@CoO催化剂的形貌表征
反应后的Pt-ZnO@CoO催化剂的形貌及元素分布情况如图8所示。由图8可以看到,反应后Co颗粒粒径明显减小,从还原后的20~40 nm减小到约12 nm,说明反应过程中Co颗粒进行了再分散。结合高角度环形暗场扫描透射电子显微镜(HAADF-STEM)和HRTEM以及EDX元素分布结果可以看到,Pt颗粒大小约为1 nm,Co物种粒径约为12 nm,两者均匀分布于ZnO载体表面,如图8(a)中的插图所示。部分Pt颗粒分布于Co物种附近,同ZnO基底形成了三元微界面。

图8 反应后Pt-ZnO@CoO催化剂的电子显微镜照片和EDX元素分布
2.6 CO2-TPD表征
为了进一步探究不同催化剂上CO2加氢性能产生差异的原因,利用CO2-TPD考察了不同催化剂的CO2吸附能力,结果如图9所示。由图9可知:3种催化剂对CO2的吸附性能有明显差别。200~300 ℃的脱附峰归属于CO2的弱吸附,350~400 ℃的脱附峰归属于CO2中等强度的吸附;Pt-ZnO@CoO催化剂的CO2吸附能力远高于ZnO@CoO,尤其CO2弱吸附的脱附峰面积明显增加,说明引入Pt之后,大幅增强了催化剂对CO2的弱吸附。这可能是Pt-ZnO@CoO催化剂具有高活性的原因之一。

图9 催化剂的CO2-TPD表征结果
2.7 CO2加氢催化反应机理初步探讨
对于Pt-ZnO@CoO催化剂,Pt通过氢溢流作用极大地促进了CoO外延层的还原,使其在ZnO表面形成金属Co颗粒,同时Pt颗粒也分散在其附近。对于CO2加氢反应,CO2解离是决速步骤,金属Co颗粒具有良好的CO2解离能力。Dong Cui等[18]报道,氢气分子可以在ZnO表面发生异裂,并溢流至Co氧化物表面。Pt与ZnO两物种的溢流效应能够加速金属Co表面的CHx物种或CHO物种进一步加氢生成CH4或CH3OH。
3 结 论
基于晶格匹配设计合成了Pt-Co-ZnO三元催化剂,通过STEM和XRD表征可知,制备的3种催化剂中Co、Pt均高度分散。Pt-ZnO@CoO催化剂还原后出现了分布均匀的Co颗粒,粒径为20~40 nm,说明Co纳米颗粒均匀分散在ZnO纳米棒表面,形成了纳米复合结构。反应过程中,Co纳米颗粒进行了再分散,粒径减小到12 nm左右,并与Pt颗粒一同分散在ZnO表面,形成三元微界面。
在固定床装置上考察了不同催化剂对CO2加氢反应的催化性能,发现Pt-ZnO@CoO催化剂对CO2加氢反应显示出优异的催化活性和稳定性。随着反应温度升高,CO2加氢转化率不断提高,CO2加氢产物中甲烷选择性不断提高,醇选择性不断降低;在反应温度为300 ℃时,CO2加氢转化率高达20.1%,产物中甲烷选择性为70.3%,醇选择性为26.3%。
结合STEM,H2-TPR,CO2-TPD表征结果,推断高密度的界面位点、较强的还原能力、较强的CO2吸附能力可能是Pt-ZnO@CoO催化剂具有高活性、高稳定性的原因。此外,基于晶格匹配的多元界面催化剂设计策略为高效CO2加氢催化剂的开发提供了新思路。
猜你喜欢
机械工业标准化与质量(2022年6期)2022-08-12
数学物理学报(2019年5期)2019-11-29
第一财经(2019年8期)2019-08-26
数学物理学报(2017年5期)2017-11-23
中国调味品(2017年2期)2017-03-20
潍坊学院学报(2016年6期)2016-04-18
哈尔滨医药(2015年2期)2015-12-01
学习月刊(2015年14期)2015-07-09
中学化学(2015年2期)2015-06-05
物理化学学报(2015年5期)2015-02-28
