
基于多信息融合的DGPMIF致病基因关联预测方法
2024-03-11 09:26马金龙翟美静
河北工业科技 2024年1期
马金龙 翟美静

摘 要:
为了解决利用单一生物数据无法揭示复杂的生物过程和疾病机制的问题,提出了一种多信息融合的DGPMIF致病基因预测方法。首先,构建一个具有疾病-表型、疾病-基因、蛋白质-蛋白质和基因-本体关联的异构网络,利用网络嵌入算法提取该异构网络中节点的低维向量表示,同时结合网络拓扑算法提取网络结构特征。其次,利用余弦相似性算法衡量节点向量的相似性,预测疾病与基因之间的关系。最后,通过对特定疾病的案例进行研究,并与经典致病基因预测方法进行对比,验证DGPMIF方法的有效性。结果表明:不同类型的关联数据对增强致病基因预测性能具有重要作用;经过多层次信息融合,提高了致病基因预测的预测性能。DGPMIF预测方法能够高效挖掘网络中蕴含的信息,对相关疾病基因关联的预测研究具有重要的参考价值。
关键词:
人工智能其他学科;致病基因;异构网络;信息融合;网络嵌入;网络结构特征
中图分类号:TP29 文献标识码:A
DOI: 10.7535/hbgykj.2024yx01004
A disease-gene association prediction method of DGPMIF based on multi-information fusion
MA Jinlong, ZHAI Meijing
(School of Information Science and Engineering, Hebei University of Science and Technology, Shijiazhuang, Hebei 050018, China)
Abstract:
In order to solve the problem of being unable to reveal complex biological processes and disease mechanisms using only a single biological data, proposed a disease-causing gene prediction method, DGPMIF, adopting a multi-information fusion strategy. Firstly, a heterogeneous network with disease-phenotype, disease-gene, protein-protein and gene-ontology associations was constructed. The network embedding algorithm was used to extract the low-dimensional vector representation of the nodes in the heterogeneous network. At the same time, the network topology algorithm was combined to extract network structural characteristics. Secondly, the cosine similarity algorithm was used to measure the similarity of node vectors and predict the relationship between diseases and genes. Finally, the effectiveness of the DGPMIF method was verified through case studies of specific diseases and comparison with classic disease-causing gene prediction methods. The results show that different types of associated data play an important role in enhancing the prediction performance of disease-causing genes, and the predictive performance of disease-causing gene prediction is improved through multi-level information fusion. DGPMIF prediction method can efficiently mine the information contained in the network, and has important reference value for prediction research on gene association of related diseases.
Keywords:
other disciplines of artificial intelligence; disease-causing genes; heterogeneous network; information fusion; network embedding; network structural characteristics
對致病基因的研究在医学研究中发挥着重要作用。在临床中,众多疾病显现出深刻而复杂的表型特征,为明确这些疾病与基因的隐秘关联,需要对候选致病基因进行精确鉴定[1]。传统方法(如连锁分析)能够确定这些基因之间的关联,但对于涉及数百甚至更多基因的复杂疾病,则成本高昂且耗时长[2]。因此,计算方法显得尤为关键。在过去的几十年里,人们利用计算方法对疾病基因之间的关联进行了大量研究。
网络表示能够简化复杂多样的生物数据,使得基于网络的方法在预测疾病基因关联方面越来越受欢迎[3]。诸多研究表明,与相同或相似疾病相关的基因通常在功能上相关,并且它们在蛋白质-蛋白质相互作用网络(PPI)中彼此相邻或接近[4]。GONZALEZ等[5]设计了一种计算致病基因的方法,观察疾病相关蛋白质之间的相互作用关系以及这些蛋白质节点在网络中的聚集倾向,利用蛋白质相互作用网络的拓扑结构来识别致病基因。PPI网络上的重启随机游走(RWR)用于预测致病基因,基于网络中随机游走过程,探索候选基因和种子基因之间的网络邻近性[6]。然而,PPI网络数据的单一性使其难以全面反映疾病与基因之间的相关信息。因此,越来越多的研究采用异构网络来解决复杂的疾病基因预测问题。相比于同构网络,异构网络在疾病基因预测方面提供了更全面、多样化和上下文感知的信息,能更好地处理生物系统的复杂性,并提供更准确的致病基因预测算法。RWRH算法是通过将RWR算法扩展到疾病基因异构网络而生成的[6-7]。VANUNU等[8]提出了基于类似异构网络的PRINCE算法,该算法可用于对所有疾病的致病基因进行全局优先排序。与此同时,XIE等[9]还提出了双随机游走(BiRW)算法来实现这一任务。基于异构数据类型,ZAKERI 等[10]提出了异构数据融合,并已被证明是可行的。异构网络与多源信息的结合可以提供多维互补的信息表示,在疾病基因预测方面比同质数据更有优势。
近年来,图嵌入方法逐渐崭露头角,成为从网络数据中挖掘有用信息的一种显著方法。这一方法也被称为网络嵌入,其旨在生成节点表示,确保在短随机步行距离内的节点拥有相近的嵌入,并能自动学习疾病和基因的潜在特征或嵌入。例如,DeepWalk[11]、Node2vec[12]和 LINE[13]在学习嵌入方面表现出了出色的性能。随后,一些研究人员通过整合新的网络嵌入技术开展了相关工作。XIANG等[14]提出了一种利用快速网络嵌入预测疾病相关基因的新方法PrGeFNE,该方法利用快速网络嵌入算法从网络中提取节点的低维表示,并重建双层异构网络。然而,如何从异构网络中提取有价值的信息来准确、快速地预测致病基因仍是当前一项具有挑战性和有意义的任务。
针对异构网络中的疾病基因预测问题,本研究提出一种创新性的多信息融合方法,即DGPMIF。首先,建立一个异构网络,涵盖多种关联,包括疾病基因关系和其他相关关联。其次,运用先进的网络嵌入算法,将这些关联转化为节点的特征表示,并融合网络结构特征。通过对多信息融合、先进的网络嵌入算法以及网络拓扑结构特征的综合运用,DGPMIF方法有望在异构网络中更全面地捕获重要特征,进一步推动疾病基因预测领域的研究进程。
1 DGPMIF方法概述
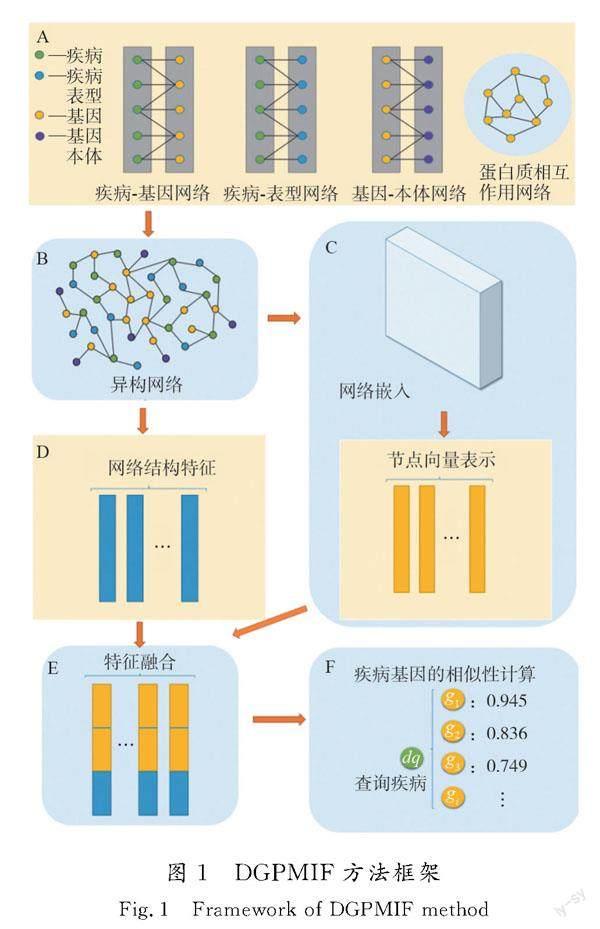
DGPMIF方法框架如图1所示。
首先,以疾病基因为核心,将来自多个信息源的数据(包括疾病基因之间的关联数据、疾病相关信息以及基因相关信息)整合到一个网络中。这一步骤的关键在于综合不同类型的信息以全面考虑各种关联性。其次,采用先进的网络嵌入算法处理这个异构网络,将其中的节点映射为低维向量表示。这些向量包含节点之间的关系信息,更准确地描述节点在网络中的位置和作用,有助于捕捉节点之间复杂的关联关系,同时,充分利用网络的拓扑结构特征。通过网络拓扑算法提取与节点在网络中的位置以及连接模式相关的信息,这一过程为节点的描述提供更多维度,能进一步丰富特征表示。最后,采用余弦相似度算法对增强的低维向量表示进行相似性计算,以量化节点之间的相似性。DGPMIF方法在异构网络中更为深入地捕获关键特征,从而提高疾病基因预测的准确性和综合性。
2 DGPMIF方法预测关键环节
2.1 构建异构网络
本文的数据集使用YANG等[15]提取的疾病基因异构网络,包括4种类型的节点:疾病、疾病表型、基因和基因本体,以及多种不同类型的相互关联关系。子网络的具体基本信息如表1所示。在获取表1中4类子网络的原始数据后,需要对其进行预处理,检查数据并删除缺失值。然后通过疾病节点和基因节点作为中间节点连接,对所有节点进行统一映射,以保证最终集成网络的准确性。本文构建的异构网络可为研究提供多种生物数据的复杂关联,有助于深入研究疾病基因的预测和相关性分析。此外,构成的网络都是无权无向图。
通过对上述疾病基因数据的分析和整合,本文重新定义了6个不同的网络,其中每个网络捕捉了不同类型的关联信息。这6个网络如下:1)疾病基因关联(DGA,简称DG);2)DGA和PPI(简称[CM(22]DGG);3)DGA和疾病表型关联(DSA)(简称[CM)]DGS);4) DGG和基因本体关联(GOA)(简称DGGG);5)DGS和GOA(简称DGSG);6) DGA,PPI,DSA,GOA (简称DGSGG)。通过构建这些网络,可以更全面地研究不同类型信息在致病基因预测中的作用,为疾病研究提供更多维度和角度。
2.2 网络嵌入算法
网络嵌入算法是在处理复杂网络中节点的低维向量时的重要方法,广泛应用于可视化、节点分类、链接预测等多种任务中。图2展示了网络嵌入算法的流程图。为了在疾病基因的复杂网络中更好地捕获和保留网络结构,本文采用了4种不同的网络嵌入算法,分别是DeepWalk[11]、Node2vec[12]、LINE[13]和 SDNE[17]。这些方法被用于提取节点向量,以更好地捕获和保留网络结构。
1)DeepWalk 是一种基于随机游走的网络嵌入算法,其通过在网络上执行随机游走模拟节点间的随机漫步过程。通过对这些随机游走序列应用Word2vec等词嵌入技术,将节点映射到一个低维向量空间中,使其在该空间中相似的节点保持相近的向量表示。
2)Node2vec 是DeepWalk的扩展,引入了参数控制随机游走策略,使得可以在節点之间平衡探索局部和全局结构。Node2vec能够更好地捕获节点的多样性和上下文信息,从而生成更具信息丰富性的节点嵌入表示。
3)LINE 是一种基于一阶和二阶邻居的网络嵌入算法。其通过最大化节点之间的一阶和二阶邻居之间的相似性学习节点的向量表示。该方法在保留网络结构信息的同时,能够有效捕获节点之间的高阶关联。
4)SDNE 是一种基于深度学习的网络嵌入算法,通过自编码器结构学习节点的嵌入表示。SDNE在保持网络的拓扑结构信息的同时,能够捕获节点之间的非线性关系,使得其在处理复杂网络时具有较强的表达能力。
在疾病基因网络中,上述算法都以将网络中的节点映射到低维向量空间为共同目标,有助于更深入地理解和分析网络的结构、以及节点之间的关系。
2.3 网络结构特征
网络的结构信息与节点属性紧密相关。常见的拓扑结构指标包括节点间的最短路径、共同邻居以及节点的度等。疾病基因网络是一个异构网络,与同构网络相比,其拥有更为丰富的结构信息。为了进一步提高预测性能,将网络的拓扑信息纳入训练样本的特征中是至关重要的。
本文对一些具有代表性的结构特征进行详细阐述,并给出其基本定义,包括度(degree,D)、度中心性(degree centrality, DC)、聚类系数(cluster coefficient,CC)、介数(betweenness,B)[18]、介数中心性(between centrality,BC)[19]、紧密中心性(closeness centrality,Cc)[20]和特征向量中心性(eigenvector centrality,EC)[21]。 对于给定的网络G=(V,E),V是节点集合,E是边的集合,用N(i)表示网络中节点i的所有邻居节点的集合,V是网络节点总数。网络中节点i的度D(i)和度中心性DC(i)定义如下:
D(i)=N(i),(1)
DC(i)=D(i)V-1。(2)
节点i的聚类系数CC(i)的定义如下:
CC(i)=2E(i)K(i)·(K(i)-1),(3)
式中:E(i)表示节点之间的边数;K(i)表示一阶邻域中的节点数。
节点的介数中心性是一种全局几何度量,能够有效反映网络中单个节点的重要性。节点i的介数中心性BC(i)见式(4)。
BC(i)=∑s≠i≠tσst(i)σst。(4)
式中:σst是从节点s到节点t的最短路径总数;σst(i)是经过节点i的路径数。
紧密中心性反映了节点与网络内其他节点的接近度。节点i的紧密中心性基于从该节点到网络中所有其他节点的平均距离di。di的倒数定义为节点i的紧密中心性Cc(i),见式(5)、式(6)。
di=1n-1∑j≠idij,(5)
Cc(i)=1di=n-1∑j≠idij,(6)
式中:n表示节点i所属的网络中的节点总数;dij表示节点i和j之间的最短距离。
一个节点的重要性不仅取决于其邻居节点的数量(即该节点的度),还取决于其邻居节点的重要性。与之相连的邻居节点越重要,则该节点就越重要。xi是节点i的重要性度量,该节点的特征向量中心性EC(i)表示为
EC(i)= [WTHX]x[WTBX]i=c∑nj≠i[WTHX]a[WTBX]ij[WTHX]x[WTBX]j,(7)
式中:c表示一个比例常数;[WTHX]a[WTBX]ij是网络的邻接矩阵。记[WTHX]x[WTBX]=[x1,x2,x3,...,xn]T,经过多次迭代达到稳态后,[WTHX]x[WTBZ]可以写成如下矩阵形式:
[WTHX]x[WTBX]=c[WTHX]Ax[WTBX],(8)
式中:[WTHX]x[WTBX]表示的是矩阵[WTHX]A[WTBX]的特征值c-1对应的特征向量。
本文整合了疾病与基因的关联数据及其相关信息,进一步增强了网络的复杂性和多样性。将网络结构特征与网络嵌入算法所得到的向量表示进行融合,作为致病基因预测的最终特征输入。鉴于网络中节点的结构特征通常以相似度值的形式体现,直接采用拼接的策略以用于特征信息的融合,进一步提升疾病相关基因预测的精度和可靠性。
2.4 余弦相似度计算
通过重建低维向量表示来测量节点对(疾病和基因)的余弦相似度。以疾病基因对的相似度计算为例,给定疾病vdx和基因vdy,N(vdx)和N(vdy)是它们的向量表示。 然后,根据余弦相似度算法,可以计算出疾病-基因对的余弦相似度,算法如式(9)所示:
cos(N(vdx),N(vdy))=cos(x,y)=x·yx·y。(9)
在应用基于向量表示的余弦相似度算法后,网络中疾病与基因对的相似度可以被准确地计算出,从而测量它们之间的相关性。再将查詢的疾病与候选基因的相关性进行排序,可以得到特定疾病的候选基因的排名列表。
3 实验与结果分析
3.1 实验环境
本文的全部实验均在Window10操作系统下完成,所有的代码均使用Python编程语言实现,并在PyCharm 集成开发环境下进行编写与调试,实验所使用的软件环境和硬件环境的相关信息分别如表2与表3所示。
3.2 设置参数
固定超参数能够确保不同网络嵌入算法之间具有可比性,并增强实验结果的稳定性,减少随机性对性能评估的影响。因此,对每个网络嵌入算法选择固定的超参数。对于DeepWalk,随机游走的步数设置为80,每个节点随机游走次数为40,窗口大小为10,嵌入维度为128;Node2vec与DeepWalk的参数设置大致相同,同时还需要考虑控制随机游走策略的2个超参数p和q,本文设置p=1.5和q=1.5;LINE方法中的参数设置包括一阶邻居和二阶邻居,采样数都设置为5,负采样率为0.5;SDNE的参数设置:隐层节点数为128,迭代次数为100次,其他参数默认。
3.3 评价指标
在评估疾病基因预测方法性能上,本文采用以下指标:准确率(accuracy,AC)、精确率(precision,PR)、召回率(recall,RE)、F1得分(F1-score,F1)和曲线下面积(area under the curve,AUC)。这些指标是评估分类器质量的常用指标,可以更全面地评估疾病基因预测方法的性能,其中F1得分通常用于综合考虑精确率和召回率,特别适合在正负样本不平衡的情况下进行评估。
3.4 疾病基因恢复实验与结果分析
为了评估网络嵌入算法在边缺失情况下的性能,将已知的边模拟为缺失,然后尝试恢复这些缺失的边。这有助于评估算法在网络重建和边预测方面的效果,以及其对网络拓扑结构的理解程度。如果网络嵌入算法能够在恢复实验中表现出色,那么其通常也能在预测任务中表现得更好。
首先,为了模拟网络中边的缺失情况,本文通过从疾病基因异构网络中随机删除一些边来实现。这些已删除的边为后续恢复实验的样本。其次,采用多种网络嵌入算法,如DeepWalk、Node2vec、LINE和SDNE,以学习网络中节点的低维向量表示。这些向量表示有助于更好地理解网络结构。最后,分别在 DG、DGG 和 DGS 网络上进行恢复实验,利用学习到的节点向量表示,试图恢复已删除的边。计算每个已删除的边样本在向量空间中的相似度分数,并将这些分数用于预测是否应该将边恢复。如果计算出的2个节点的相似度的值大于0.5,则将边恢复;若低于0.5,则视为2个节点无关联。这一系列实验评估了不同算法在恢复任务上的性能,从而更深入地了解它们在疾病基因网络中的表现。每种算法的恢复性能如表4所示,每种算法在同一网络上的最佳性能以粗体标记。由表4可知,Node2vec 算法对边的恢复性能最好。因此,本文后续实验都是基于Node2vec算法。此外,使用DGS和DGG网络算法比使用DG网络算法表现更好,这表明考虑更多信息后(例如疾病表型关联或PPI网络),可以提高疾病基因关联的恢复性能。
3.5 致病基因预测实验与结果分析
为了提高疾病基因的预测性能,本文选择与疾病或基因紧密相关的数据源进行融合,例如PPI、基因本体和疾病表型。然而,不适当的数据融合也会导致不利影响。因此,研究来自不同数据源的信息融合如何影响疾病基因预测方法的性能很有必要。
通过采用疾病基因及其相关数据的各种组合方式,本研究构建了6种不同的网络(DG、DGG、DGS、DGGG、DGSG 和 DGSGG),并将这些网络用作实验数据集。由于预测阶段主要针对疾病与基因之间的相关性,因此在提取训练样本的过程中,随机选择了疾病基因子网络中50%的连边,将它们作为正样本,并从原网络中移除了这些选定的连边。处理后的网络用于接下来的特征提取环节。首先,在预测阶段,随机抽取一定数量的负样本,正负样本的比例为1∶1。其次,为所有提取的样本分配相应的标签。最后,采用五折交叉验证的方法,将数据集划分为5个子集。每次实验,其中4个子集用于训练模型,而剩余的1个子集用于测试。为了确保实验的可靠性和鲁棒性,更准确地评估DGPMIF方法在预测任务中的性能,实验重复5次,每次使用不同的子集作为测试集。最终,取5次实验结果的平均值作为最终的性能评估结果。此外,其他子网不会被处理。获得4类节点(疾病、基因、疾病表型、基因本体)的向量表示以及对应的结构特征后,根据节点类型进行拼接,作为测试样本的特征表示。为了结果的一致性,将结构特征拼接在疾病或基因节点向量表示的后面。
表5展示了使用每个网络的疾病基因预测方法的性能,并用粗体文本标记了所有网络中的最佳性能。
由表5可知,相较于DG网络,DGG网络的AUC增加了0.017 1,而DGS网络的AUC增加了0.019 3,表现出更优的预测表现。然而,在DGG网络和DGS网络的基础上,
整合基因本体信息的DGGG网络和DGSG网络的改进效果并不显著。同时,DGSGG网络的AUC低于除DG网络之外的其他4个网络。显然,纳入更多信息的DGSGG网络并没有显著改善预测结果,这表明疾病表型关联和PPI网络信息之间可能存在干扰。此外,在所有实验结果中,融合网络结构特征的DGSG(DGSG+SF)网络(AUC:0.954 1,AC:90.21%,F1:0.913 2,PR:0.923 8,RE:0.902 9)取得了最佳性能。在同一网络中,融合网络结构特征的评价指标优于仅使用向量表示。这说明网络的结构特征能够增强网络嵌入算法得到的向量表示,对提升预测效果具有积极作用。
3.6 与其他方法对比实验与结果分析
所提出的DGPMIF方法通过融合网络结构特征增强低维向量表示来预测潜在的致病基因。为了验证此方法的优越性,通过引入RWRH[6]、RWR[7]、BiRW[8]、PRINCE[8]和CIPHER[22]5种经典算法进行比较。而在致病基因预测中,通常会有大量的候选基因,但实际上只有其中的一小部分是真正的致病基因。为了提高预测的精确性,将每种疾病相关的基因排名列表获得后,选择前k个基因(TOP@k)作為候选基因,其中k分别取值为3、5、10,并使用精确度和召回率作为评价标准。
融合网络结构特征的DGPMIF方法与其他方法的预测结果详见表6。通过考察前k个候选基因的精确率和召回率可知,融合网络特征算法在这2个评估指标上的表现均优于未融合网络特征的算法。对比5种经典基线方法发现,RWRH展现了最佳性能。从表6中可以看到,当k设定为3时,RWRH的PR值为0.323 4,RE值达到0.546 9。采用DGG网络的预测性能低于RWRH,但DGG+SF网络的预测性能高于RWRH,表明网络特征的融合可以在一定程度上提高疾病基因预测的性能。采用DGSG+SF网络取得了最好的性能,与DG网络相比,PR@3和RE@3分别提高了0.108 5和0.239 3,表明选择与疾病或基因密切相关的数据进行有效整合能够增强疾病基因预测。将其与RWRH相比,PR@3和RE@3分别提高了0.105 0和0.142 8,表明与经典算法相比,多信息融合方法在疾病基因预测方面展现出了更为优异的表现。综合来看,无论k取何值,几种典型方法的预测性能均低于融合网络特征的多信息融合方法。因此,DGPMIF方法确实能够有效提升预测潜在致病基因的性能。
3.7 案例研究与结果分析
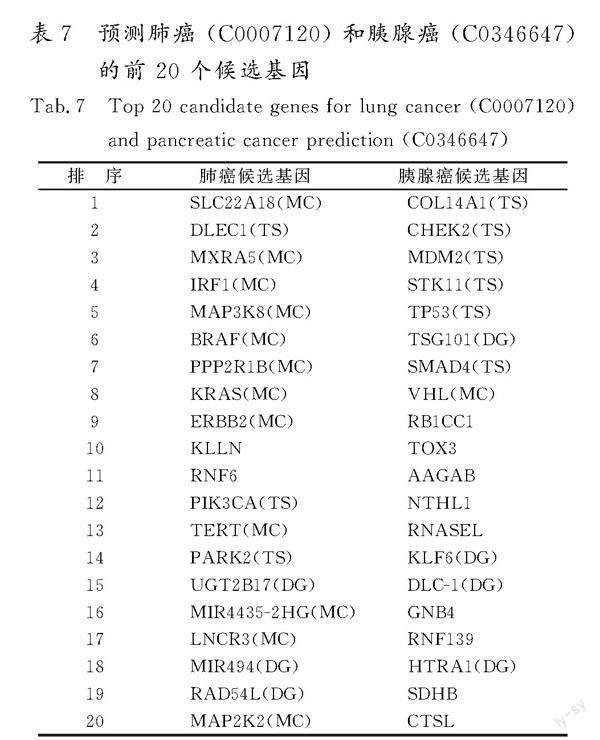
通过计算某种疾病与数据集中所有基因之间的相关性得分,得到此疾病的候选基因排名列表。为了阐明生物学意义,使用 DGSG+SF网络评估了DGPMIF对肺癌 (C0007120)和胰腺癌 (C0346647)这2种疾病的预测效果。这2种疾病的前20个预测基因如表7所示。首先,从数据集中筛选出这2种疾病已知的关联基因,以此作为标记数据进行模型训练。其次,模型训练完毕后,将其应用于数据集中剩余的未标记基因数据,计算这些基因与特定疾病的关联概率,进而对未知关联基因进行排序。为验证模型的预测效果,参考了MalaCards在线生物数据库,并查阅了相关的科学文献,核实预测出的候选基因与特定疾病是否存在已知关联。
就肺癌而言,前 20 个候选基因中的 DLEC1(排序为2)、PIK3CA(排序为12) 和 PARK2 (排序为14)是数据集中的已知基因(标记为 TS)。此外,在MalaCards数据库中,SLC22A18(排序為1)、MXRA5(排序为3)、IRF1(排序为4)、MAP3K8(排序为5)、BRAF(排序为6)、PPP2R1B(排序为7)、KRAS (排序为8)、ERBB2 (排序为9)、TERT (排序为13)、MIR4435-2HG (排序为16)、LNCR3 (排序为17) 和 MAP2K2 (排序为20) 是已知的肺癌基因(标记为MC)。为了全面评估候选基因,对已发表的生物医学文献进行检索以进行验证。排除未验证的KLLN(排序为10)和RNF6(排序为11),剩余3个基因UGT2B17(排序为15)、MIR494(排序为18)和RAD54L(排序为19)(标记为DG)可以得到佐证并有相应的文献证据。GALLAGHER等[23]指出,UGT2B17的缺失与女性的肺癌风险显著增加有关。此外,文献[24]也提到了MIR494与非小细胞肺癌的相关性。
与此同时,有研究进一步揭示了RAD54L在肺癌进展中所扮演的角色[25]。
此外,文献[26]揭示了TSG101在胰腺癌的发生和进展中的核心作用。有研究强调了KLF6通过上调转录因子3 (ATF3)的激活从而抑制胰腺癌进展的机制[27]。文献[28]指出了DLC-1可能在胰腺癌的致病机制中扮演关键角色。文献[29]进一步印证了胰腺癌与HTRA1之间的联系。这些研究表明,DGPMIF方法预测出的新基因在很大程度上与特定疾病真正相关,从而为多信息融合策略的有效性提供了进一步的支撑。
4 结 语
本文提出的DGPMIF方法,通过构建疾病基因异构网络,整合更多与疾病或基因相关的信息,应用融合网络结构特征的方式,致力于解决异构网络中致病基因的预测问题。
1)DGPMIF方法聚合了多个重要的信息源,创建了一个综合网络,涵盖了疾病与基因的关联信息以及其他相关信息,从而深度挖掘和理解了疾病与基因之间的复杂关系。
2)DGPMIF方法不仅仅局限于单一层面的网络分析,而是通过融合多层面的网络结构特征,准确捕捉和呈现了网络中节点间的关系。这一多层面特征融合方法使得模型更加精准地理解了网络的动态和复杂性,为研究提供了新的思路和可能性。
DGPMIF方法在疾病预测中取得了优异成绩,结合其他生物医学特征,如药物靶点网络、组织特异性网络和基因表达等,或许能够进一步增强其预测能力。这是未来研究的新方向和研究重点。
参考文献/References:
[1]
HINDORFF L A,SETHUPATHY P,JUNKINS H A,et al.Potential etiologic and functional implications of genome-wide association loci for human diseases and traits[J].Proceedings of the National Academy of Sciences of the United States of America,2009,106(23):9362-9367.
[2] VASIGHIZAKER A,JALILI S.C-PUGP:A cluster-based positive unlabeled learning method for disease gene prediction and prioritization[J].Computational Biology and Chemistry,2018,76:23-31.
[3] ZHANG Yan,XIANG Ju,TANG Liang,et al.Pgagp:Predicting pathogenic genes based on adaptive network embedding algorithm[J].Frontiers in Genetics13,2022,13:1087784.
[4] BARABASI A L,GULBAHCE N,LOSCALZO J.Network medicine:A network-based approach to human disease[J].Nature Reviews Genetics,2011,12(1):56-68.
[5] GONZALEZ M W,KANN M G.Protein interactions and disease[J].PLoS Computational Biology,2012,8(12):e1002819.
[6] LI Yongjin,JAGDISH C P.Genome-wide inferring gene-pheno-type relationship by walking on the heterogeneous network[J].Bioinformatics,2010,26(9):1219-1224.
[7] KHLER S,SEBASTIAN B,DENISE H,et al.Walking the interactome for prioritization of candidate disease genes[J].The American Journal of Human Genetics,2008,82(4):949-958.
[HJ1.9mm]
[8] VANUNU O,MAGGER O,RUPPIN E,et al.Associating genes and protein complexes with disease via network propagation[J].PLoS Computational Biology,2010,6(1):e1000641.
[9] XIE Maoqiang,HWANG T,RUI K.Prioritizing disease genes by bi-random walk[C]// Knowledge Discovery and Data Mining.Berlin:Springer,2012:292-302.
[10]ZAKERI P,ELSHAL S,MOREAU Y.Gene prioritization through geometric-inspired kernel data fusion[C]//2015 IEEE International Conference on Bioinformatics and Biomedicine (BIBM).Washington:IEEE,2015:1559-1565.
[11]PEROZZI B,AL-RFOU R,SKIENA S.DeepWalk:Online learning of social representations[C]//In Proceedings of the 20th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining.New York:Association for computing machinery,2014:701-710.
[12]GROVER A,LESKOVEC J.Node2vec:Scalable feature lear-ning for networks[C]// KDD ′16:Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining.[S.l.]:[s.n.],2016:855-864.
[13]TANG Jian,QU Meng,WANG Mingzhe,et al.LINE:Large-scale information network embedding[C]// Proceedings of the 24th International Conference on World Wide Web.Florence:International World Wide Web Conferences Steering Committee,2015:1067-1077.
[14]XIANG Ju,ZHANG Ningrui,ZHANG Jiashuai,et al.PrGeFNE:Predicting disease-related genes by fast network embedding[J].Methods,2021,192:3-12.
[15]YANG Kuo,WANG Ruyu,LIU Guangming,et al.HerGePred:Heterogeneous network embedding representation for disease gene prediction[J].IEEE Journal of Biomedical and Health Informatics,2019,23(4):1805-1815.
[16]MENCHE J,SHARMA A,KITSAK M,et al.Disease networks[J].Uncovering Disease-disease Relationships Through the Incomplete Interactome.Science,2015,347(6224):1257601.
[17]WANG Daixin,PENG Cui,ZHU Wenwu.Structural deep network embedding[C]//In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining.[S.l]:ACM,2016:1225-1234.
[18]GOH K I,OH E,KAHNG B,et al.Betweenness centrality correlation in social networks[J].Physical Review E,2003,67(1/2):017101.
[19]BARTHELEMY M.Betweenness centrality in large complex networks[J].The European Physical Journal.B,2004,38(2):163-168.
[20]SALAVATI C,ABDOLLAHPOURI A,MANBARI Z.Ranking nodes in complex networks based on local structure and improving closeness centrality[J].Neurocomputing,2019,336:36-45.
[21]BONACICH P.Some unique properties of eigenvector centra-lity[J].Social Networks,2007,29(4):555-564.
[22]WU X B,JIANG R,ZHANG M Q,et al.Network-based global inference of human disease genes[J].Molecular Systems Biology,2008,4:189.
[23]GALLAGHER C J,MUSCAT J E,HICKS A N,et al.The UDP-glucuronosyltransferase 2B17 gene deletion polymorphism:Sex-specific association with urinary 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol glucuronidation phenotype and risk for lung cancer[J].Cancer Epidemiology Biomarkers & Prevention,2007,16(4):823-828.
[24]LU Bing,LYU Hong,YANG Zhiqiang,et al.LncRNA PCAT29 up-regulates the expression of PTEN by down-regulating miR-494 in non-small-cell lung cancer to suppress tumor progression[J].Critical Reviews in Eukaryotic Gene Expre-ssion,2021,31(6):9-15.
[25]LIU Changjiang,REN Wei,ZHANG Zhixin,et al.DNA repair/recombination protein 54L promotes the progression of lung adenocarcinoma by activating mTORC1 pathway[J].Human Cell,2023,36(1):421-433.
[26]ZHU Yufu,XU Yang,CHEN Tianze,et al.TSG101 promotes the proliferation,migration,and invasion of human glioma cells by regulating the AKT/GSK3 β/β-Catenin and RhoC/cofilin pathways[J].Molecular Neurobiology,2021,58(5):2118-2132.
[27]XIONG Qunli,ZHANG Zhiwei.YANG Yang,et al.Krüppel-like factor 6 suppresses the progression of pancreatic cancer by upregulating activating transcription factor 3[J].Journal of Clinical Medicine,2023,12(1):200.
[28]ZHENG Zhenjiang,TAN Chunlu,XIANG Guangming,et al.Deleted in liver cancer-1 inhibits cell growth and tumorigenicity in human pancreatic cancer[J].Oncology Letters,2013,6(2):521-524.
[29]CHENG Hao,ZHU Hao,CAO Meng,et al.HtrA1 suppresses the growth of pancreatic cancer cells by modulating Notch-1 expression[J].Brazilian Journal of Medical and Biological Research,2018,52(1):e7718.
收稿日期:2023-09-09;修回日期:2023-12-26;責任编辑:王淑霞
基金项目:河北省省级科技计划资助项目(23550801D)
第一作者简介:
马金龙(1981—),男,河北定州人,副教授,博士,主要从事生物信息学方面的研究。
E-mail:mzjinlong@163.com
马金龙,翟美静.基于多信息融合的DGPMIF致病基因关联预测方法
[J].河北工业科技,2024,41(1):27-35.
MA Jinlong, ZHAI Meijing. A disease-gene association prediction method of DGPMIF based on multi-information fusion
[J]. Hebei Journal of Industrial Science and Technology,2024,41(1):27-35.
猜你喜欢
黄河之声(2022年10期)2022-09-27
中学生数理化(高中版.高二数学)(2022年4期)2022-05-25
中学生数理化(高中版.高二数学)(2022年4期)2022-05-25
小学教学研究(2022年5期)2022-04-28
新世纪智能(数学备考)(2021年9期)2021-11-24
当代陕西(2019年15期)2019-09-02
学苑创造·A版(2018年11期)2018-02-01
中学生数理化·八年级物理人教版(2017年11期)2017-04-18
中国洗涤用品工业(2017年2期)2017-04-16
读者(2017年5期)2017-02-15
