
Au n(n=2,3,4)团簇与乙醇分子相互作用的第一性原理研究*
2011-10-23 01:23于永江杨传路安义鹏王华阳
物理学报 2011年2期
于永江 杨传路 安义鹏 王华阳
(鲁东大学物理学院,烟台 264025)
(2010年2月4日收到;2010年3月12日收到修改稿)
Aun(n=2,3,4)团簇与乙醇分子相互作用的第一性原理研究*
于永江†杨传路 安义鹏 王华阳
(鲁东大学物理学院,烟台 264025)
(2010年2月4日收到;2010年3月12日收到修改稿)
利用密度泛函理论研究了Aun(n=2,3,4)团簇与乙醇分子的吸附机理.研究结果表明:Aun(n=2,3,4)团簇分别能够吸附1到n个乙醇分子,生成Aun-(C2H6O)1-n配合物;Au4团簇吸附乙醇分子有多种构型,通过分析吸附能和Mulliken电荷分布,确定了4个乙醇分子的吸附顺序及相应的稳定构型;Aun(n=3,4)团簇在吸附最后一个乙醇分子时改变了前面Au—O成键的作用模式,而是选择Au—H成键;作为吸附主体的金团簇和被吸附的乙醇分子在吸附前后构型变化都很少,它们之间的吸附作用为弱相互作用.
金团簇,乙醇分子,密度泛函理论
PACS:31.15.es,21.60.Gx
1.引 言
过渡金属团簇特别是金团簇,由于其独特的物理和化学性质,近年来引起人们的研究兴趣.纳米尺度的金团簇及其化合物已被广泛地应用于催化反应、材料吸附和光的吸收中,因而理解金团簇和纳米粒子的最稳定构型和电子性质具有重要的理论意义和应用背景[1—6].对于较小的Aun团簇(n<10),人们对其结构的认识已达成共识,认为金团簇形成以三角形为基础的平面二维结构[7].对于纯金团簇研究在实验和理论方面已有大量报道.实验上对金团簇的研究主要集中在离子团簇上,通过测定其迁移速率[8]和光电子能谱[9]获得有关的结构信息.由于实验条件和测量技术的限制,对中性金团簇的实验研究比较困难,人们对其结构和性能的认识主要依赖理论研究.
最近关于由分子和金的纳米微粒形成的配合物的光学、电子和催化性质在实验和理论方面研究引起了人们的广泛兴趣.分子被吸附在金的表面、金团簇或者金的纳米微粒上已经被人们广泛的研究,例如苯硫醇、硫醇盐、丙酮、二乙基的酮、乙醛、烷烃、平面的分子噻吩等有机分子已见研究报道[10—14],对于无机分子,例如 O2和 CO 也有相应的理 论 和 实 验 研 究 报 道[15,16]. 李 迎 春 等[17]研 究 了Aun+团簇与甲醇分子的相互作用,发现多个CH3OH分子可以同时被吸附在团簇上,提供了一个金团簇和CH3OH分子之间相互作用的更加清晰的图景.乙醇作为有机溶质,在工业、医疗和生活方面应用广泛,目前尚未见金团簇和多个乙醇分子之间相互作用的研究报道,本文在前期工作的基础上,采用密度泛函方法研究小的 Aun(n=2,3,4)团簇对乙醇分子的吸附作用,从理论上对金纳米微粒与乙醇的相互作用提供更丰富的信息.
2.理论方法
本文中的所有结果都是利用密度泛函软件Materials Studio中的 DMol3模块[18]进行计算得到.DMol3使用原子数值函数作为原子基函数.数值基组可以正确表示体系的电荷分布,对电子极化效应进行精确的描述.我们选用程序内置的带有极化方程的双数值基组(DNP),金是重金属元素,相对论效应明显,计算中采用全电子相对论选项.DNP基组的大小与Gauss函数杂化的B3LYP/6-31G相仿,但要比之更加精确.这种高精度的数值基组可以减少基组重叠效应,对体系进行准确的描述.DFT交换相关势函数采用广义梯度矫正(GGA)的(perdew-burke-emzerhof,PBE)函数[19],从而避免了局域密度近似(LDA)对原子间键长的低估和对能量的不正确估算.
我们在几何优化中选用的收敛标准是:能量1.0 × 10-6Hartree(Ha)(1 Ha=27.2114 eV),力常数4.5×10-4Ha/和步长0.002.自洽场的收敛标准是电子密度均方根的变化量小于1×10-5/3.为了确保得到的能量构型都是势能面上的极小值,而不是过渡态或是亚稳态,我们对所有最低能量构型都作了频率分析,所有计算结果没有虚频,确认为稳定结构.
3.结果与讨论
密度泛函及相应收敛标准已经在文献[17]中进行了讨论.在研究金团簇和乙醇分子的相互作用之前,我们首先研究了金团簇 Aun(n=2,3,4)的几何结构.
3.1.Au n(n=2,3,4)团簇的平衡几何结构

图 1 Au n(n=2,3,4)团簇的结构构型 (a)n=2,(b1)—(b3)n=3,(c1)—(c5)n=4
图1中分别给出了 Aun(n=2,3,4)团簇的所有可能的空间几何构型.根据前面所述的方法,分别优化了各个几何构型,计算得到了其点群对称性结构、能量和平均键长等参数性质,并根据总能量大小分别判断了Aun(n=2,3,4)团簇的最稳定几何结构.团簇的结构和性质参数如表1中所示.显然Au2团簇只有1种几何结构构型,其点群对称性为D∞h(a),能量值为 - 36795.8988 Ha,HOMO-LUMO能隙(Gap)为1.763 eV,键长为2.489,振动频率为184.8 cm-1,与实验键长2.47和振动频率191 cm-1符合的较好[20];研究发现 Au3团簇共有三种无虚频的稳定结构构型,其点群结构分别为 D∞h(b1),C2v(b2)和 D3h(b3).其中具有等边三角形形状的结构具有最小能量,能量为 -55193.8672 Ha,键长为2.620,HOMO-LUMO能隙为1.342 eV;对于Au4团簇,分别优化了可能的五种几何构型,并通过能量判断得知Au4团簇最稳定构型是平面菱形D2h对称性结构,是由腰长为 2.386,底边长为2.547的两个等腰三角形构成,其能量为-73591.8741 Ha,HOMO-LUMO 能隙为 1.018 eV.这与以前的报道中得到的金团簇的最稳定构型相符[21,22].
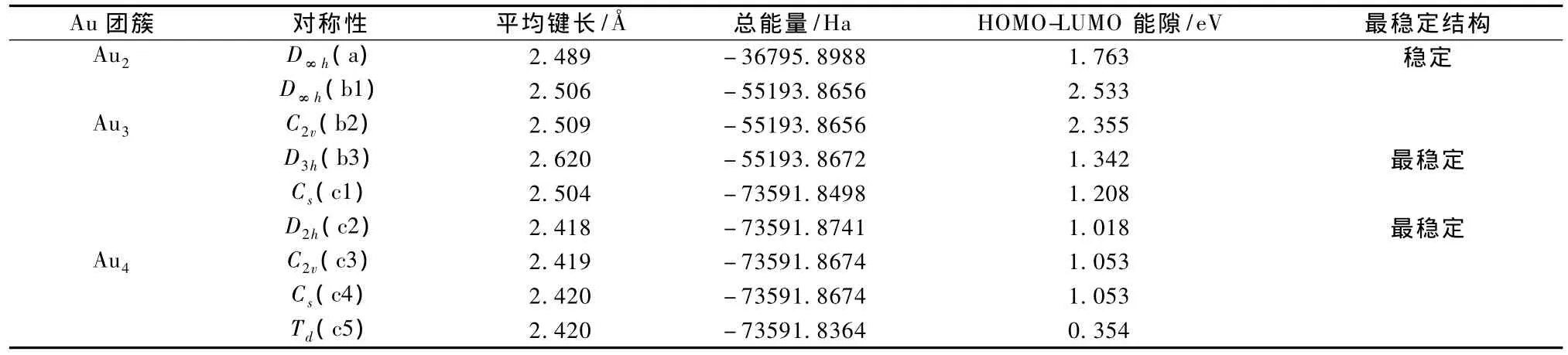
表1 从头算得到的Au n(n=2,3,4)团簇的结构性质参数
3.2.Au n(n=2,3,4)团簇和乙醇分子的相互作用
Shafai等[23]研究了 Aun(n=3,5,7,9,13)团簇和单个丙酮分子的相互作用,他们指出,当金原子或者金二聚物与丙酮分子的羰基氧相互作用的时候得到最大的结合能(BE),相比于C和H而言,Au更容易与O结合而相互成键,所以我们所选的初始构型即为Au—O吸附接触构型.图2中(a)和(b)展示了Au2团簇吸附乙醇分子的数目从1到2个的情况.由于Au2的两个原子的顶点是等价的,所以吸附相同个数的分子只有一种结构模型.图3中(a)—(c)展示了Au3团簇吸附乙醇分子的数目从1到3个的情况.由于等边三角形Au3的三个原子也是等价的,所以吸附相同个数的分子也只有一种结构模型.图中黑色球表示氧原子,灰色球表示碳原子,浅灰色球表示金原子,白色球表示氢原子.

图2 Au2团簇吸附乙醇分子 (a)吸附1个乙醇分子,(b)吸附2个乙醇分子
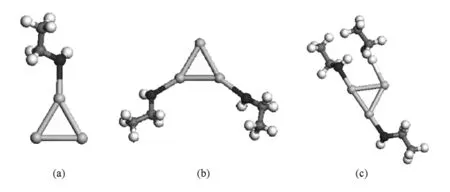
图3 Au3团簇吸附乙醇分子 (a)吸附1个乙醇分子,(b)吸附2个乙醇分子,(c)吸附3个乙醇分子
Au4团簇和乙醇分子的相互作用如图4中所示,我们给出了Au4团簇分别吸附1到4个乙醇分子所有可能的八种结构,分别进行结构优化并得到了其无虚频的稳定结构.
我们分别给出了Aun(n=2,3,4)团簇与乙醇分子吸附作用后形成的配合物的结构性质和参数信息如表2所示.其中一个重要的性质参数是金团簇与乙醇分子之间的吸附能Eb,以Au3团簇吸附3个乙醇分子为例,Eb可用下式表示:
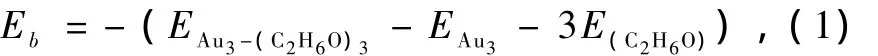
其中,EAu3为Au3团簇的总能量,E(C2H6O)为乙醇分子的总能量,3为吸附乙醇分子的个数,EAu3-(C2H6O)3为吸附后构成的Au3—(C2H6O)3配合物的总能量,吸附能为正值说明吸附是稳定的.
3.3.讨论
1)分子结构分析讨论.研究发现 Aun(n=2,3,4)团簇HOMO-LUMO能隙值随着n的增加而减少,可见当Aun(n=2,3,4)团簇随着 n的增加,化学活性逐步提高.
通过Au2O优化计算,得出Au—O成键结果,键长为1.933,配合物Aun(C2H6O)n的Au—O键长介于2.181—2.576之间,与其基本符合,可以断定 Aun(C2H6O)n上 是 Au—O 成 键.另 外,Aun(C2H6O)n配合物的吸附能都为正值,说明它们已经形成了稳定结构.
对于乙醇分子,其吸附前后的结构略有变化,其中C—C键长略有减小,C—O键长略有增加.随着吸附乙醇分子个数的增加,Au—O键长变化不明显,但对于Au3和Au4团簇各自吸附的第3个和第4个乙醇分子时,Au—O之间距离较大,变为3.295,而Au—H之间距离各是2.303和2.310,再通过DMol3中成键名称分析发现它们已经形成了Au—H键,这类似于 Kryachko等[24]在研究 DNA碱基与金团簇的相互作用时发现了一种Au—H键.但上述的第3个特别是第4个乙醇分子结构几乎没有变化,可以断定它们的相互作用比较弱.如图5中(a)—(c)所示,进一步研究了Au3团簇和1个乙醇分子通过Au—H成键进行相互作用,发现也会产生稳定结构,平均吸附能为0.895 eV,比 Au—O成键情况的吸附能少了约三分之一,可见 Au—H键比Au—O键作用要弱.研究发现配合物成键具有方向性,Au—O键方向性明显,它和 Aun团簇在一个平面内,但Au—H键与Aun团簇不在一个平面内.

图4 Au4团簇吸附乙醇分子 (a1),(a2)为吸附1个乙醇分子的2种结构,(b1)—(b3)为吸附2个乙醇分子的3种结构,(c1)—(c2)为吸附3个乙醇分子的2种结构,(d)为吸附4个乙醇分子的结构

图5 Au3团簇吸附乙醇分子通过不同的氢原子Au-H成键
对于Au2团簇,吸附后 Au—Au键长缩短而且吸附2个乙醇时 Au—Au键长变得更短.对于 Au3团簇的变化则是,吸附1个乙醇分子时Au3团簇的键长均略有减少,超过1个乙醇分子时Au3团簇的键长均略有增加,且随着吸附个数的增加而略有增加.
2)电子布局分析Au—H键形成的原因.Au3团簇吸附2个乙醇分子后,空位上的金原子Mulliken电荷为-0.262e,由于所带负电荷较大,因此第三个乙醇分子与其相互作用时Au—O成键就比较困难,而形成了Au—H键.根据金原子上的Mulliken电荷分布,可以分析Au4团簇吸附乙醇分子的先后顺序(a2)—(b2)—(c2)—(d),(c2)构型上的空位金原子Mulliken电荷为-0.312e,带负电较大,再跟第4个乙醇分子相互作用时,不易形成 Au—O而出现Au—H成键.假如从(c1)构型向(d)发展,可以理解为受第4个乙醇分子的影响,原来吸附的一个乙醇分子受其影响,发生了位置变化,Au—O键消失,Au—H键形成.

表2 Au n—(C2 H6 O)n配合物的结构性质参数
3)结构和稳定性分析.对于Au4团簇和乙醇分子作用前后,吸附一个乙醇分子的情况有两种构型,如图4(a1)和(a2)所示.根据吸附能的大小可知,两种构型的吸附能相差近1倍,可以说Au4团簇吸附一个乙醇分子时(a2)构型可能性较大.而对于吸附两个分子的情况则有三种吸附构型,如图4中(b1),(b2)和(b3)所示.根据吸附能的大小可知此时图4(b2)结构的吸附能最大,是Au4团簇吸附两个乙醇分子时的最稳定构型.对于吸附三个乙醇分子时的两种情况而言,计算所得的吸附能(c2)较大,更易成为配合物构型.对于吸附四个乙醇分子的情况,此时金团簇的键长已有较大的增加,在乙醇分子的相互作用下,Au4团簇被拉伸,面积略有增大.
整体而言,Aun(n=2,3,4)团簇与乙醇分子相互作用的吸附能较少,乙醇分子结构未发生较大变化,即未出现C—O、C—C或C—H键的断裂,Aun(n=2,3,4)团簇结构和键长变化不明显,团簇中的Au—O键比Au2O原子之间形成的键要长,可以认为金团簇和乙醇分子之间的相互作用是一种弱相互作用.
4.结 论
本文利用密度泛函理论研究了小的Aun(n=2,3,4)团簇与乙醇分子的相互作用.首先通过优化确定了 Aun(n=2,3,4)团簇的最稳定构型分别为D∞h,D3h和D2h对称性的二维结构.然后在此基础上研究了Aun(n=2,3,4)团簇与1—n个乙醇分子的相互作用.研究发现:Au2团簇能够分别吸附1—2个乙醇分子,Au3团簇能够分别吸附1—3个乙醇分子,且这种吸附作用较弱.Au4团簇与1—4个乙醇分子有多种吸附构型,通过吸附能和Mulliken电荷分布我们判断了分别吸附1到4个乙醇分子稳定构型的先后顺序.Aun(n=2,3,4)团簇和乙醇分子之间相互作用后,各自结构没有明显变化,均为弱相互作用.金团簇与乙醇分子构成的配合物中,Au—O键具有方向性,它们都跟金团簇处在同一个平面中.Aun(n=3,4)团簇在吸附最后一个乙醇分子改变了前面Au—O成键的作用模式,而是Au—H成键,且相互作用更弱.为研究金团簇的性质提供了更丰富的信息.
[1]Valden M,Lai X,Goodman D W 1998Science281 1647
[2]Wang C,Zhao R N,Han JG 2006J.Chem.Phys.124 194301
[3]Liu L,Zhao R N,Han JG,Liu F Y,Pan G Q,Sheng L S 2009J.Phys.Chem.A 113 360
[4]Fang F,Jiang G,Wang H Y 2006ActaPhys.Sin.55 2241(in Chinese)[方 芳、蒋 刚、王红艳2006物理学报 55 2241]
[5]Zhao L X,Feng X J,Cao T T,Liang X,Luo Y H 2009Chin.Phy.B 18 2709
[6]Gu J,Wang S Y,Gou B C 2009ActaPhys.Sin.58 3338(in Chinese)[顾 娟、王山鹰、苟秉聪 2009物理学报 58 3338]
[7]Han Z,Zhang D J,Liu C B 2009ActaChim.Sin.67 387(in Chinese)[韩 哲、张东菊、刘成卜 2009化学学报 67 387]
[8]Furche F,Ahlrichs R,Weis P,Jacob C,Gilb S 2002J.Chem.Phys.117 6982
[9]Hakkinen H,Yoon B,Landman U,Li X,Zhai H J,Wang L S 2003J.Phys.Chem.A 107 6168
[10]Lee T H,Ervin K M 1994J.Phys.Chem.98 10023
[11]Sara L,Fabrizio C 2004J.Chem.Phys.120 10062
[12]Dietrich G,Krückeberg S,Lützenkirchen K,Schweikhard L,Walther C 2000J.Chem.Phys.112 752
[13]Shafai G S,Sharan S,Sailaja K,Vaishali S,Kanhere D G 2007J.Chem.Phys.126 014704
[14]Lavrich D J,Wetterer S M,Bernasek S L,Scoles G 1998J.Phys.Chem.B 102 3456
[15]Varganov SA,Olson R M,Gordon M S,Mills G,Metiu H 2002J.Chem.Phys.119 2531
[16]Hayashi T,Tanaka K,Haruta M 1998J.Catal.178 566
[17]Li Y C,Yang C L,Sun M Y,Li X X,An Y P,Wang M S,Ma X G,Wang D H 2009J.Phys.Chem.A 113 1353-1359
[18]Perdew J,Burke P K,Ernzerhof M 1996Phys.Rev.Lett.77 3865
[19]Delley B 2006J.Phys.Chem.A 110 13632
[20]Bishea G A,Morse M D 1991J.Chem.Phys.95 5646
[21]Deka A,Deka R C 2008J.Mol.Stru.:Theochem870 83
[22]Mao H P,Wang H Y,Ni Y,Xu G L,Ma M Z,Zhu ZH,Tang Y J 2004ActaPhys.Sin.53 1766(in Chinese)[毛华平、王红艳、倪 羽、徐国亮、马美仲、朱正和、唐永坚 2004物理学报53 1766]
[23]Shafai G S,Sharan S,Sailaja K,Vaishali S,Kanhere D G 2007J.Chem.Phys.126 014704
[24]Kryachko E S,Remacle F 2005J.Phys.Chem.B 109 22746
PACS:31.15.es,21.60.Gx
First principles study on the interaction of Aun(n=2,3,4)clusters w ith ethanolmolecules*
Yu Yong-Jiang†Yang Chuan-Lu An Yi-Peng Wang Hua-Yang
(School of Physics,Ludong University,Yantai 264025,China)
(Received 4 February 2010;revised manuscript received 12 March 2010)
The mechanism of the adsorption between Aun(n=2,3,4)clusters and ethanol molecules is investigated with the density functional theory.It is found that Aun(n=2,3,4)clusters can adsorb several ethanolmolecules to form the Aun-(C2H6O)1-ncompounds.There are many adsorbing conformations when the ethanol molecules are adsorbed by the Au4cluster.The adsorbing sequence and the corresponding stable configurations are confirmed through the analysis of the adsorption energies and the Mulliken electrical displacement.When the last ethanol molecule is adsorbed,the bonding style changes from the Au—O bond to the Au—H bond.The constructions of the Aunclusters and the ethanolmolecule are less changed in the adsorbing process.And the interaction between Aunclusters and ethanol molecules is weak interaction.
Au cluster,ethanol,density functional theory
*山东省自然科学基金(批准号:Z2008A02)资助的课题.
*Project supported by the Natural Science Foundation of Shandong Province,China(Grant No.Z2008A02).
猜你喜欢
辽宁科技大学学报(2022年5期)2023-01-04
山西大学学报(自然科学版)(2022年5期)2022-11-23
军民两用技术与产品(2022年1期)2022-06-01
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
原子与分子物理学报(2020年5期)2020-03-17
青岛大学学报(工程技术版)(2019年2期)2019-09-10
考试周刊(2018年39期)2018-04-19
北京航空航天大学学报(2017年10期)2017-04-20
枣庄学院学报(2015年5期)2016-01-09
中学化学(2015年8期)2015-12-29
