
贵金属原子与点缺陷石墨烯的键增强作用
2012-12-05 02:27解鹏洋庄桂林吕永安王建国李小年
物理化学学报 2012年2期
解鹏洋 庄桂林 吕永安 王建国 李小年
(浙江工业大学化学工程与材料学院,杭州310014)
贵金属原子与点缺陷石墨烯的键增强作用
解鹏洋 庄桂林*吕永安 王建国*李小年
(浙江工业大学化学工程与材料学院,杭州310014)
通过密度泛函理论研究了Ag、Au、Pt原子在完美和点缺陷(包括N掺杂、B掺杂、空位点缺陷)石墨烯上的吸附以及这些体系的界面性质.研究表明Ag、Au不能在完美的石墨烯上吸附,N、B掺杂增强了三种金属与石墨烯之间的相互作用.而空位点缺陷诱发三种金属在石墨烯上具有强化学吸附作用.通过电子结构分析发现,N掺杂增强了Au、Pt与C形成的共价键,而Au、Ag与B形成了化学键.空位点缺陷不仅是金属原子的几何固定点,同时也增加了金属原子和碳原子之间的成键.增强贵金属原子和石墨烯相互作用的顺序是:空位点缺陷>>B掺杂>N掺杂.
密度泛函理论; 石墨烯;金;铂;银
1 Introduction
Graphene,as an emerging material,has attracted tremendous attention in different research fields since 2004.1-9Noble metal nanoparticles10-13are of great interests due to their unique catalytic properties.Metal nanoparticles supported on graphene nanocomposites,14-20feature the characteristics of both grahpene and metal nanoparticles,particularly notable because they not only inherit their intrinsic properties but also extract some unique cooperative properties,which exhibit promising applications in nanobiotechnology,nanoelectronics,energy storage,catalysis,etc.Therefore,understanding of the interaction between graphene and metal nanoparticles is the first step to realize these applications.21Meanwhile,the interaction between metal adatoms/clusters and graphene also depends on the preparation methods and determines the properties of the formed nanocomposites.
For metal clusters/low dimensional carbon(such as carbon nanotubes,graphene,fullerene),chemical or physical methods are commonly used to prepare metal/carbon nanocomposities.22-28For the chemical methods,organic compounds or functional groups25,28,29adsorbed on the surface of carbon materials can serve as the anchoring site of these metal clusters.Therefore,the metal nanoparticles adhere to the graphene via these“linkers”.The metal/graphene nanocomposites are generally prepared by depositing metal particles on graphene/graphite oxide23or the chemical functionlized graphene30-36sheets.The physical methods are to grow or deposit metal nanoparticles directly onto the carbon nanotubes(CNTs)or grahite/graphene surface via electron beam evaporation,37or thermal evaporation.38,39At present,few experimental studies have been reported on the preparation of metal/graphene nanocomposites with the physical methods.Independent of the preparation methods, the ultimate purpose of these methods is to modify the inert properties of pristine CNTs/grahene,which can be attributed to two types of modifications.One is to modify the properties by the surface species,36and the other is to substitute the lattice carbon with foreign elements33,40-47or form various vacancies.48-50It is well-known that N,B atoms are the only two foreign elements incorporated into an sp2carbon network of CNTs42,51-54or graphene55,56without significantly affecting their geometric structures.
Several theoretical studies57-62have been conducted on the interaction between metal adatoms or small clusters and the pristine graphene or the graphene with vacancies.These studies show the ionic bonding for metal adatoms of groups I-III elements and covalent bonding for transition metal atoms with d valence electrons,noble metals,and group IV elements58on the pristine graphene.The very weak interaction between Ag,Au and graphene is identified from the previous study.57
To the best of our knowledge,no systematic theoretical studies of noble metal adatoms or clusters on the point defected, which include B-,N-doping and a single vacancy defect,graphene have been reported.In this study,we investigated the interactions between three typical noble metal adatoms(Ag,Au, and Pt)and point defected graphene by means of density functional theory(DFT)calculations,which is further compared with these on the pristine graphene.
2 Calculation methods
All calculations were carried out under the generalized gradient approximation(GGA)with the Perdew-Burke-Ernzerhof (PBE)63functional,within a plane wave-pseudopotential scheme,by using the PWSCF package in Quantum ESPRESSO.64The ultrasoft pseudopotentials65were used to describe electron-ion interactions.The kinetic energy cutoffs for the smooth part of the electronic wave function and the augmented electron density were 25 and 200 Ry(1 Ry=13.6056923 eV), respectively.In this study,by using the(6,6)graphene,the point-defect concentration is about 2.7% (molar fraction), which represents realistic experimental conditions.The pristine,N-,B-doping graphene,and graphene with vacancies are termed as Gr,N-Gr,B-Gr,and vac-Gr,respectively.The Brillouin zone integration was performed with the k points generated for 6×6×1 Monkhorst-Pack grid,66which were convergent by using 8×8×1,10×10×1,and 12×12×1 Monkhorst-Pack grids.All the atoms involved in calculations were fully relaxed until each component of the residual force on each atom was smaller than 0.3 eV·nm-1.
The binding energy(Eb)of metal adatom on the graphene was typically calculated as follows:

where EM1,EGr,and E(M1+Gr)represent the energies of the most stable gas phase metal adatom,the graphene,and the combined systems of metal adatom and graphene,respectively.
3 Results and discussion
3.1 Electronic properties of graphene
In order to investigate the effect of point defect on the electronic properties of graphene,the band structures,density of states(DOS),and charge differences were calculated,as shown in Fig.1.Inspecting of the band structures and DOS of Fig.1,it can be found that the electronic bands of B-,N-doped graphene feature similar dispersive characteristics to that of pristine one,but both of the Fermi levels are shifted up by-0.57 and+0.53 eV.The obtained band gaps of B-and N-Gr are still zero.Therefore,the B-and N-Gr can be attributed to p-type and n-type semiconductors,comparable to those in the reported studies.67On the other hand,vac-Gr shows different electronic characteristics from others,in which the band gap rises from zero to 0.77 eV.This may be due to that the broken sp2configuration in vac-Gr induces the impurity states consisting of dangling sp3orbitals of carbons,which slightly shift upto conduction band.The charge density differenceswhere the doping is the boron,nitrogen,and carbon of B-,N-Gr and pristine graphene;orwhere c2 is the 2-coordinate carbon in vac-Gr,respectively)induced by the B-or N-doping and vacancies are also shown in Fig.1. The red and blue colors represent the electron accumulation and depletion,respectively.It can be seen that the B,N show the electron deficiency and accumulation.And B-,N-doping also induce the charge redistribution on the graphene,which can be confirmed from Löwdin analysis.The charges of B and N are 0.12e and-0.02e,which is consistent with the analysis of DOS.On the pristine graphene,the charge is uniformly distributed on the carbon atoms.While for vac-Gr,the charge is depleted around the carbon vacancies.
3.2 Binding of metal adatoms on grapheme
As the reference systems,we also investigated the adhesion of Ag,Au,and Pt adatoms on the pristine graphene.Due to the very similar adhesion properties of Ag and Au,Fig.2 only shows the binding energies and optimized configurations of the most and the second stable structures of Au and Pt on the investigated graphene.We found that Ag(Eb=0.02 eV)and Au(Eb= 0.20 eV)adatoms are very weakly bound on the pristine graphene,which are in agreement with the available literature.68The geometries of graphene have no obvious changes after the adsorption of Au and Ag.In contrast,Pt shows much stronger adhesion properties.The most and the second stable binding sites are the bridge of two carbons(Eb=1.90 eV)and a top of one carbon(Eb=1.76 eV),respectively.It can be seen that the weak bonding between metal clusters(especially Ag and Au) and graphene must be strengthened in order to utilize these composited nanomaterials.In this study,two kinds of methods, including removing one carbon and substituting one carbon by the B,N elements,have been taken into account.
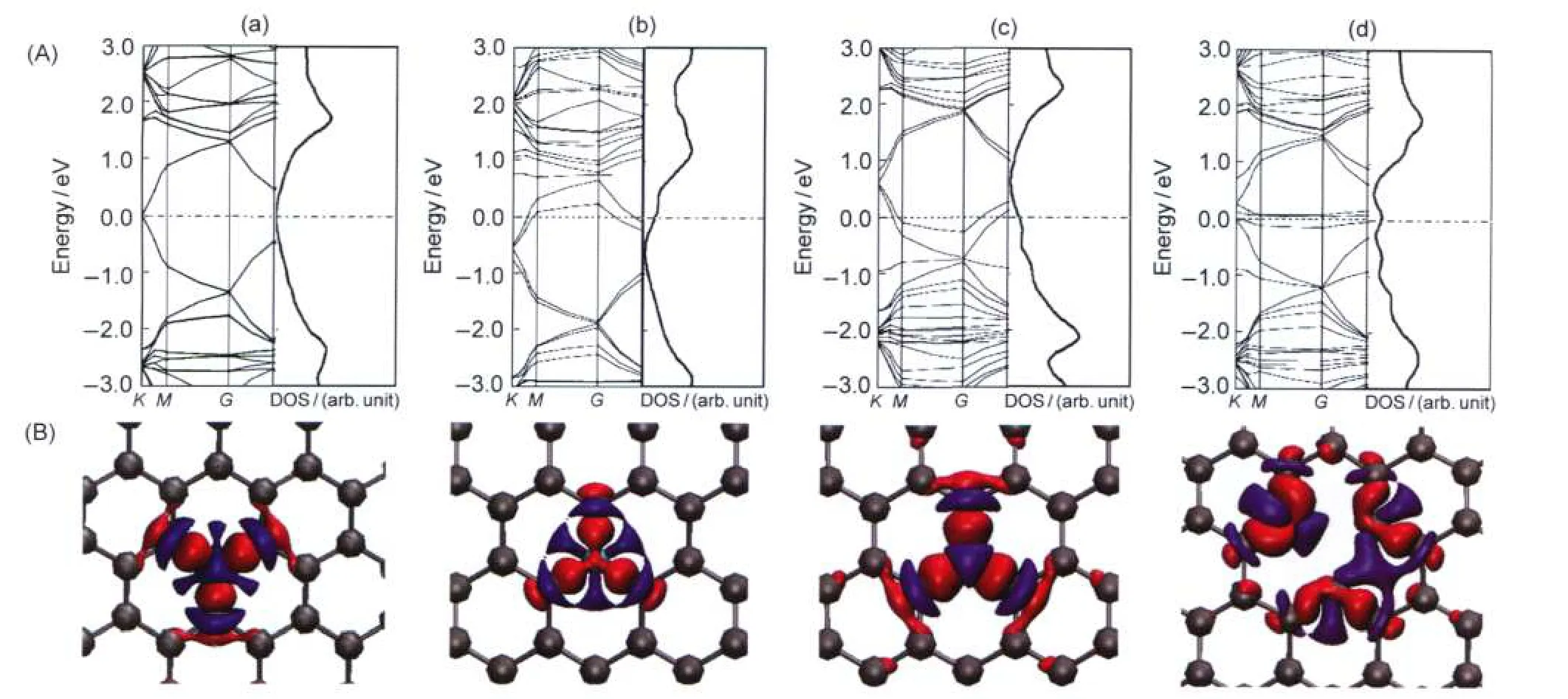
Fig.1 Band structures and total density of states(A)and charge density differences(B)of(a)Gr,(b)N-Gr,(c)B-Gr,and(d)vac-Gr

Fig.2 Optimized geometries and binding energies(in the parentheses)of the most and second stable structures ofAu and Pt adatoms on(a)Gr,(b)N-Gr,(c)B-Gr,and(d)vac-Gr
3.3 Binding of metal adatoms on N-,B-,vac-Gr
It can be seen that the weak bonding between Au,Ag and pristine graphene is caused by the inert electronic properties of graphene.On the other hand,the electronic properties of graphene can be modified by the B-,N-doping and vacancies.In this section,we further investigated the adhesion and binding of noble metal adatoms on the B-,N-,and vac-Gr.For N-Gr, the most favorable binding site is the top of o-carbon atoms (ortho-carbon)rather than nitrogen,in which the binding energies are 0.13 and 0.84 eV for Ag and Au adatoms,respectively.For Au adatoms,the physisorption on the pristine graphene turns to weak chemisorption on the N-Gr,in which the distances between Au and carbon are 0.320 and 0.224 nm.The most and the second stable binding sites of Pt adatoms on N-Gr are both bridge of two carbon atoms.The most favorable binding site of Pt1on N-Gr is the bridge of o-and p-carbon atoms(paracarbon)of N.The second favorable binding site of Pt1on N-Gr is the bridge of p-and m-carbon(meta-carbon)atoms of N. The binding energies are 2.25 and 2.05 eV for the most and the second Pt adatoms,which are about 0.35 and 0.15 eV larger than that on pristine graphene,respectively.For Ag and Au on B-Gr,the most favorable binding sites are both top of boron atoms,in which the binding energies are 1.11 and 1.29 eV,re-spectively.For Pt on B-Gr,the most and the second favorable sites prefer the bridge of B and o-carbon and the top of B with the binding energies of 2.65 and 2.43 eV,respectively.On vac-Gr,the most favorable binding sites of metal adatoms are all located directly at the vacancies,in which metal adatoms bond with three carbon atoms.The corresponding bond lengths of M―C(M:metal adatoms)are 0.230,0.209,and 0.194 nm for Ag,Au,and Pt adatoms,respectively.The binding energies of Ag,Au,and Pt adatoms are 1.80,2.36,and 7.53 eV,respectively,which increase at least about four times larger than those on pristine graphene.
It is observed that the binding energies for the most and the second stable noble metal adatoms on N-,B-,and vac-Gr increase compared with that on the pristine graphene(Fig.2). Moreover,the initial geometries on the different sites of N-, B-,and vac-Gr(as depicted in Fig.3(b))were taken into account,and the resulting binding energies are shown in Fig.3(a). Firstly,we found that the binding energies of these metal adatoms are nearly same when they are located at the forth carbon away from the B,N,and vacancies,which is nearly the same as that on the pristine graphene.Secondly,on the B-and vac-Gra,Au and Pt always move back into the favorable binding site,even when they are initially located at the sites slightly away from the B-and vacancy about two carbons.
The most stable binding energies of the three kinds of noble metal on four different types of graphene are shown in Fig.4.It can be seen that N-,B-doping,and vacancy enhance bonding between metal adatoms and graphene.The enhanced role increases according to this order:N-doping,B-doping,and vacancy.For Ag and Au,the weak physisorption on the pristine graphene becomes the chemisorption by these modifications. Especially,the point defects(vacancies)increase the binding energies of the three kinds of metal adatoms at least four times larger than that on the pristine graphene.
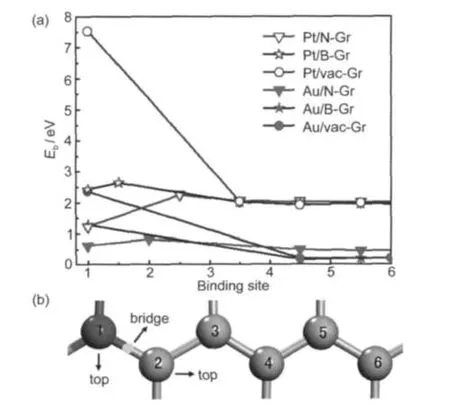
Fig.3 (a)Binding energy ofAu and Pt adatoms on the different sites of N-Gr,B-Gr,and vac-Gr;(b)illustration of different binding sites of N-Gr,B-Gr,and vac-Gr

Fig.4 Binding energy of the most stableAg,Au,and Pt adatoms on the Gr,N-Gr,B-Gr,and vac-Gr
3.4 Different mechanisms to enhance the bonding of metal adatoms on N-,B-,vac-Gr
The interactions between three noble metal adatoms and graphene can be enhanced by the B-,N-doping and point-defected carbon vacancy.But the most favorable sites and the enhanced degree are very different.Therefore,it is necessary to investigate the binding mechanisms of the three kinds of metal adatoms on different forms of graphene.
Projected density of states(PDOS)of metal adatoms and the atoms directly bonded with metal on pristine,N-Gr,B-Gr,and graphene with vacancies are shown in Fig.5.For the case of pristine graphene,there is no overlap between Au and carbon, while some hybridization between p band of carbon and d band of Pt is found at 1.8 eV above Fermi level.Furthermore, inspection of the PDOS of Au or Pt/N-Gr reveals that the dangling 2p bands of nitrogen anchor at the vicinity of Fermi level.Compared with that on pristine graphene,there is little influence of nitrogen on the PDOS of Pt and carbon.While nitrogen induces some hybridization between carbon p orbital and Au d orbital.It is interesting to observe that there is no overlap between the bands of boron and Au at Fermi level,while the 2p band of boron at Fermi level can overlap with the d band of Pt. It may be explained that the binding energy of Pt/B-Gr is larger than that of Au/B-Gr.In addition,scrutinizing PDOS of Au or Pt/vac-Gr can find that strong hybridization between p band of the carbon and d band of Au or Pt exists on the vac-Gr,resulting in the strong adhesion of metal adatoms.The PDOS differences of Au and Pt/vac-Gr at the Fermi level lead to larger binding energy of Pt/vac-Gr than that of Au/vac-Gr.Generally, it can be concluded that(1)doping N atom,B atom or vacancy defect acting as anchoring site can effectively enhance the interaction between Au or Pt and graphene;(2)among three types of adsorption case,Pt exhibits much stronger interaction with doped graphene than that ofAu.
The charge density differences induced by the adsorbed metal adatoms on pristine graphene,N-Gr,B-Gr,and graphene with vacancy are shown in Fig.6.We find that Ag adatoms have very similar properties to Au ones.Therefore,only the charge differences of Au/graphene and Pt/graphene system have been shown in Fig.6.Firstly,there are no charge transfers between Au and pristine graphene.The covalent bond is formed between Pt and carbon of pristine graphene.Secondly, N-doping does not change the bond characters between Pt and graphene,but enhances the interaction a little bitter.However, for Au,N-,B-doping plays a more important role than Pt.The covalent bond between Au and o-carbon of N is formed on N-Gr.The bond between Au and B is formed on B-Gr.The formation of these bonds changes the adhesion properties of Au on graphene,which results from the very weak physisorption to moderate chemisorption of Au.Thirdly,the role of vacancies on the enhanced bonding between metal adatoms and graphene is much stronger than N-,B-doping.It is observed that vacancies are not only the geometrically anchoring site but also the electron redistribution sites.However,the N-,B-doping mainly only induces the electron redistribution within the graphene.

Fig.5 Projected density of states of metal(d orbital)and the bonded atoms(carbon,nitrogen,or boron) (p orbital)in metal adatoms/grapheneZero mark is Fermi level.

Fig.6 Charge density differences of metal/graphene induced byAu and Pt adatomsThe red and blue colors represent for the electron accumulation and depletion,respectively.
4 Conclusions
By means of density functional theory calculations,our study demonstrates that the adhesion of noble metal(Au,Ag, and Pt)adatoms on the graphene can be enhanced either by the N-,B-doping or by the vacancies.For the same metal,the enhanced role in the binding energy increases in this order: N-doping,B-doping,and vacancies.The N-,B-doping leads to the enhancement of the covalent bond between Au and carbon atoms and formation of the chemical bond between Au or Ag and B,respectively.While point vacancies mainly act as the geometrically anchoring sites of metal adatoms and the electron reservoir.On the same graphene,the binding energies of the three kinds of metal adatoms increase in this order:Ag,Au, and Pt.The enhanced bonding between noble metal clusters and graphene will play a vital role in the application of noble metal clusters/graphene composite materials.
(1) Novoselov,K.S.;Geim,A.K.;Morozov,S.V.;Jiang,D.; Zhang,Y.;Dubonos,S.V.;Grigorieva,I.V.;Firsov,A.A. Science 2004,306,666.
(2) Li,H.;Ma,X.Y.;Dong,J.;Qian,W.P.Anal.Chem.2009,81, 8916.
(3) Li,Y.F.;Zhou,Z.;Shen,P.W.;Chen,Z.F.ACS Nano 2009,3, 1952.
(4) Saha,B.;Shindo,S.;Irle,S.;Morokuma,K.ACS Nano 2009,3, 2241.
(5) Xu,X.L.;Zhou,G.L.;Li,H.X.;Liu,Q.;Zhang,S.;Kong,J. L.Talanta 2009,78,26.
(6)Yang,Y.H.;Sun,H.J.;Peng,T.J.;Huang,Q.Acta Phys.-Chim. Sin.2011,27,736.[杨勇辉,孙红娟,彭同江,黄 桥.物理化学学报,2011,27,736.]
(7) Hu,Y.J.;Jin,J.;Zhang,H.;Wu,P.;Cai,C.X.Acta Phys.-Chim.Sin.2010,26,2073.[胡耀娟,金 娟,张 卉,吴 萍,蔡称心.物理化学学报,2010,26,2073.]
(8) Xu,N.;Kong,F.J.;Wang,Y.Z.Acta Phys.-Chim.Sin.2011, 27,559.[徐 宁,孔凡杰,王延宗.物理化学学报,2011,27, 559.]
(9) Sun,D.L.;Peng,S.L.;Ouyang,J.;Ouyang,F.P.Acta Phys.-Chim.Sin.2011,27,1103.[孙大立,彭盛霖,欧阳俊,欧阳方平.物理化学学报,2011,27,1103.]
(10) Zhang,J.;Sasaki,K.;Sutter,E.;Adzic,R.R.Science 2007,315, 220.
(11)Yoon,B.;Hakkinen,H.;Landman,U.;Worz,A.S.;Antonietti, J.M.;Abbet,S.;Judai,K.;Heiz,U.Science 2005,307,403.
(12) Matthey,D.;Wang,J.G.;Wendt,S.;Matthiesen,J.;Schaub,R.; Laegsgaard,E.;Hammer,B.;Besenbacher,F.Science 2007, 315,1692.
(13) DeVries,G.A.;Brunnbauer,M.;Hu,Y.;Jackson,A.M.;Long, B.;Neltner,B.T.;Uzun,O.;Wunsch,B.H.;Stellacci,F. Science 2007,315,358.
(14) Park,S.;Lee,K.S.;Bozoklu,G.;Cai,W.;Nguyen,S.T.;Ruoff, R.S.ACS Nano 2008,2,572
(15) Lightcap,I.V.;Kosel,T.H.;Kamat,P.V.Nano Lett.2010,10, 577.
(16)Li,B.;Lu,G.;Zhou,X.Z.;Cao,X.H.;Boey,F.;Zhang,H. Langmuir 2009,25,10455.
(17) Klusek,Z.;Dabrowski,P.;Kowalczyk,P.;Kozlowski,W.; Olejniczak,W.;Blake,P.;Szybowicz,M.;Runka,T.Appl.Phys. Lett.2009,95,113114.
(18) Li,Y.X.;Wei,Z.D.;Zhao,Q.L.;Ding,W.;Zhang,Q.;Chen,S. G.Acta Phys.-Chim.Sin.2011,27,858.[李云霞,魏子栋,赵巧玲,丁 炜,张 骞,陈四国.物理化学学报,2011,27,858.]
(19) Wu,X.Q.;Zong,R.L.;Mu,H.J.;Zhu,Y.F.Acta Phys.-Chim. Sin.2010,26,3002.[吴小琴,宗瑞隆,牟豪杰,朱永法.物理化学学报,2010,26,3002.]
(20)Wen,Z.L.;Yang,S.D.;Song,Q.J.;Hao,L.;Zhang,X.G.Acta Phys.-Chim.Sin.2010,26,1570.[温祝亮,杨苏东,宋启军,郝 亮,张校刚.物理化学学报,2010,26,1570.]
(21) Sutter,P.;Hybertsen,M.S.;Sadowski,J.T.;Sutter,E.Nano Lett.2009,9,2654.
(22)Xu,C.;Wang,X.;Zhu,J.W.J.Phys.Chem.C 2008,112,19841.
(23) Jasuja,K.;Berry,V.ACS Nano 2009,3,2358.
(24) Fullam,S.;Cottell,D.;Rensmo,H.;Fitzmaurice,D.Adv.Mater. 2000,12,1430.
(25) Carrillo,A.;Swartz,J.A.;Gamba,J.M.;Kane,R.S.; Chakrapani,N.;Wei,B.Q.;Ajayan,P.M.Nano Lett.2003,3, 1437.
(26) Li,J.;Moskovits,M.;Haslett,T.L.Chem.Mater.1998,10, 1963.
(27)Azamian,B.R.;Coleman,K.S.;Davis,J.J.;Hanson,N.; Green,M.L.H.Chem.Commun.2002,366.
(28) Marsh,D.H.;Rance,G.A.;Whitby,R.J.;Giustiniano,F.; Khlobystov,A.N.J.Mater.Chem.2008,18,2249.
(29)Liu,L.;Wang,T.X.;Li,J.X.;Guo,Z.X.;Dai,L.M.;Zhang, D.Q.;Zhu,D.B.Chem.Phys.Lett.2003,367,747.
(30) Li,J.;Liu,C.Y.Eur.J.Inorg.Chem.2010,8,1244.
(31) Pasricha,R.;Gupta,S.;Srivastava,A.K.Small 2009,5,2253.
(32) Shen,J.F.;Shi,M.;Li,N.;Yan,B.;Ma,H.W.;Hu,Y.Z.;Ye, M.X.Nano Res.2010,3,339.
(33)Wen,Y.Q.;Xing,F.F.;He,S.J.;Song,S.P.;Wang,L.H.; Long,Y.T.;Li,D.;Fan,C.H.Chem.Commun.2010,46,2596.
(34) Liu,S.;Wang,J.Q.;Zeng,J.;Ou,J.F.;Li,Z.P.;Liu,X.H.; Yang,S.R.J.Power Sources 2010,195,4628.
(35)Liu,W.C.;Lin,H.K.;Chen,Y.L.;Lee,C.Y.;Chiu,H.T.ACS Nano 2010,4,4149.
(36)Kim,Y.K.;Na,H.K.;Min,D.H.Langmuir 2010,26,13065.
(37) Zhang,Y.;Franklin,N.W.;Chen,R.J.;Dai,H.J.Chem.Phys. Lett.2000,331,35.
(38) Gingery,D.;Buhlmann,P.Carbon 2008,46,1966.
(39) Bittencourt,C.;Felten,A.;Douhard,B.;Ghijsen,J.;Johnson,R. L.;Drube,W.;Pireaux,J.J.Chem.Phys.2006,328,385.
(40)Wei,D.C.;Liu,Y.Q.;Wang,Y.;Zhang,H.L.;Huang,L.P.;Yu, G.Nano Lett.2009,9,1752.
(41)Ghosh,K.;Kumar,M.;Maruyama,T.;Ando,Y.J.Mater.Chem. 2010,20,4128.
(42) Liang,Y.X.;Shui,M.;Li,R.S.Acta Phys.-Chim.Sin.2007, 23,1647.[梁云霄,水 淼,李榕生.物理化学学报,2007,23, 1647.]
(43) Chi,M.;Zhao,Y.P.Comp.Mater.Sci.2009,46,1085.
(44) Kang,J.;Deng,H.X.;Li,S.S.;Li,J.B.J.Phys.:Condens. Matter 2011,23,346001.
(45) Jung,N.;Kim,B.;Crowther,A.C.;Kim,N.;Nuckolls,C.; Brus,L.ACS Nano 2011,5,5708.
(46)Lv,Y.A.;Zhuang,G.L.;Wang,J.G.;Jia,Y.B.;Xie,Q.Phys. Chem.Chem.Phys.2011,13,12472.
(47) Geng,D.S.;Yang,S.L.;Zhang,Y.;Yang,J.L.;Liu,J.;Li,R. Y.;Sham,T.K.;Sun,X.L.;Ye,S.Y.;Knights,S.Appl.Surf. Sci.2011,257,9193.
(48) Carlsson,J.M.;Hanke,F.;Linic,S.;Scheffler,M.Phys.Rev. Lett.2009,102,166104.
(49) Jack,R.;Sen,D.;Buehler,M.J.J.Comput.Theor.Nanos.2010, 7,354.
(50) Palacios,J.J.;Fernandez-Rossier,J.;Brey,L.Phys.Rev.B 2008,77,195428.
(51) Liu,X.M.;Romero,H.E.;Gutierrez,H.R.;Adu,K.;Eklund,P. C.Nano Lett.2008,8,2613.
(52) Williams,Q.L.;Liu,X.;Walters,W.;Zhou,J.G.;Edwards,T. Y.;Smith,F.L.Appl.Phys.Lett.2007,91,143116.
(53)Lv,Y.A.;Cui,Y.H.;Xiang,Y.Z.;Wang,J.G.;Li,X.N.Comp. Mater.Sci.2010,48,621.
(54) Lee,D.H.;Lee,W.J.;Kim,S.O.Nano Lett.2009,9,1427.
(55) Late,D.J.;Ghosh,A.;Subrahmanyam,K.S.;Panchakarla,L. S.;Krupanidhi,S.B.;Rao,C.N.R.Solid State Commun.2010, 150,734.
(56)Dai,X.Q.;Li,Y.H.;Zhao,J.H.;Tang,Y.N.Acta Phys.-Chim. Sin.2011,27,369.[戴宪起,李艳慧,赵建华,唐亚楠.物理化学学报,2011,27,369.]
(57)Hu,L.B.;Hu,X.R.;Wu,X.B.;Du,C.L.;Dai,Y.C.;Deng,J. B.Phys.B-Condens.Matter 2010,405,3337.
(58) Chan,K.T.;Neaton,J.B.;Cohen,M.L.Phys.Rev.B 2008,77, 235430.
(59) Boukhvalov,D.W.;Katsnelson,M.I.Appl.Phys.Lett.2009, 95,023109.
(60)Akturk,O.U.;Tomak,M.Phys.Rev.B 2009,80,085417
(61) Valencia,H.;Gil,A.;Frapper,G.J.Phys.Chem.C 2010,114, 14141.
(62) Rodriguez-Manzo,J.A.;Cretu,O.;Banhart,F.ACS Nano 2010, 4,3422.
(63) Perdew,J.P.;Burke,K.;Ernzerhof,M.Phys.Rev.B 1996,77, 3865.
(64) Giannozzi,P.;Baroni,S.;Bonini,N.;et al.J.Phys.:Condens. Matter 2009,21,395502.
(65) Vanderbilt,D.Phys.Rev.B 1990,41,7892.
(66) Monkhorst,H.J.;Pack,J.D.Phys.Rev.B 1976,13,5188.
(67) Huang,B.Phys.Lett.A 2011,375,845.
(68)Wang,J.G.;Lv,Y.A.;Li,X.N.;Dong,M.D.J.Phys.Chem.C 2009,113,890.
July 22,2011;Revised:November 1,2011;Published on Web:November 2,2011.
Enhanced Bonding between Noble Metal Adatoms and Graphene with Point Defects
XIE Peng-Yang ZHUANG Gui-Lin*LU¨Yong-An WANG Jian-Guo*LI Xiao-Nian
(College of Chemical Engineering and Materials Science,Zhejiang University of Technology,Hangzhou 310014,P.R.China)
The adhesion of Ag,Au,and Pt adatoms on pristine graphene and that containing point defects including N-substitution,B-substitution,and a single vacancy,as well as the interfacial properties of these systems,were investigated using density functional theory.The calculations show that Ag and Au cannot bind to pristine graphene.In contrast,B and N-doping increase the interaction between Ag,Au,or Pt metal adatoms and graphene,while a vacancy defect leads to the strong chemisorption of metal adatoms on graphene.Based on electronic structural analysis,N-doping strengthens the covalent bond between Au or Pt and carbon atoms,while B-doping leads to the formation of a chemical bond between Au or Ag and B. The vacancy defect acts as an anchoring site for metal adatoms and increases the bonding between metal adatoms and carbon atoms.Therefore,three types of point defect can effectively enhance the interaction between noble metal adatoms and graphene in the sequence:vacancy defect>>B-doping>N-doping.
Density functional theory;Graphene;Au;Pt;Ag
10.3866/PKU.WHXB201111021www.whxb.pku.edu.cn
*Corresponding authors.WANG Jian-Guo,Email:jgw@zjut.edu.cn.ZHUANG Gui-Lin,Email:glzhuang@zjut.edu.cn;Tel:+86-571-88871037. The project was supported by the National Natural Science Foundation of China(20906081).
国家自然科学基金(20906081)资助项目
O641
猜你喜欢
原子与分子物理学报(2020年5期)2020-03-17
中国金属通报(2019年9期)2019-10-21
原子能科学技术(2019年6期)2019-06-14
陶瓷学报(2019年5期)2019-01-12
物理化学学报(2018年6期)2018-03-08
物理化学学报(2015年5期)2015-02-28
读者欣赏(2014年6期)2014-07-03
语文知识(2014年2期)2014-02-28
物理化学学报(2013年1期)2013-07-25
有色金属加工(2012年5期)2012-07-28
