
5-硝氨四唑类金属起爆药的量子化学研究
2013-07-07 12:27盛涤伦朱雅红陈利魁解战峰
火工品 2013年3期
仝 辉,盛涤伦,朱雅红,陈利魁,杨 斌,解战峰
(陕西应用物理化学研究所,陕西 西安,710061)
硝氨四唑是四唑衍生物中很重要的一类化合物,具有很高的N含量(大于64.6%),且其芳香性结构导致其有很好的动力学和热力学稳定性;同时,5-NATz分子中含有能量很高的N环骨架且含氧,有很好的氧平衡;其密度、生成焓和气体生成量都较高,气体产物多为氮气,可达到少烟或无烟的效果;其离子盐可作为新型环保起爆药替代叠氮化铅[1-4]。
笔者在BLYP/DND水平下对4种5-硝氨四唑进行SCF计算,首次给出了它们的全优化几何构型、电子结构、IR 和不同温度下的热力学性质,并对计算结果进行了分析和讨论。
1 计算方法
本研究运用美国 Accelrys公司的 Materials Studio软件包的Dmol3模块(密度泛函理论)BLYP/DND计算4种5-硝氨四唑金属起爆药的结构和性质,获得全优化分子几何构型、电荷分布等若干量化参数。全部分子初始构型均由Materials Studio软件包的Visualizer模块获得,然后对分子进行几何构型全优化和频率计算,收敛精度取程序内定值,所得结构均为势能面上的极小点(无虚频)。以Dmol3模块计算分子的有关分子结构、电子结构、原子间键级、分子总能量、前线分子轨道能级及其差值、红外光谱和热力学等性质。
2 结果讨论
2.1 分子几何构型
起爆药是受外界能量的触发而自身发生剧烈化学反应的化学物质,其本质是分子中化学键的断裂,其分子结构中所含基团的稳定性对起爆药感度等性质有着重要的影响。在常规单质起爆药分子中,大都含有各种不稳定的基团,而它们的性质、数量及所在位置决定着该起爆药的感度。
本文所研究的化合物为: NaNAT(1)、CaNAT(2)、Cu(NAT)2(3)、Cu(1-mel-NAT)2(4)。图 1 为分子优化几何构型示意图。
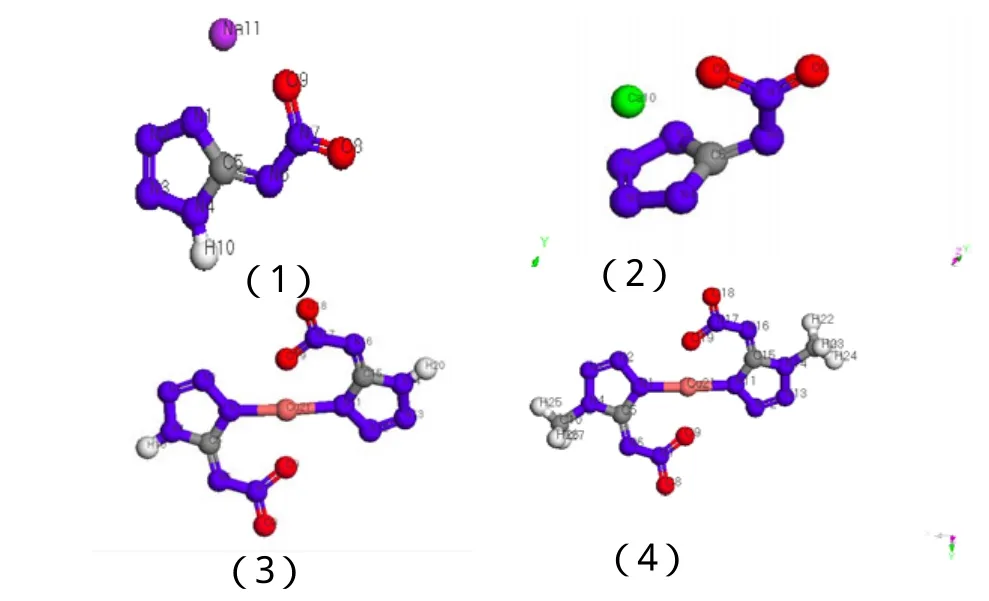
图1 BLYP/DNP优化几何构型示意图Fig.1 The optimized geometry structure by BLYP/DNP method
2.1.1 键长
键长是反映化合物结构的重要参数之一,它对分子的结构有直接影响,还对分子的能量及偶极矩等性质产生影响。表1为BLYP/DNP优化几何键长。表2为理论键长与计算键长的比较。分析表2可知:Na、Ca以共价键形式与硝氨四唑环结合。(3)和(4)的Cu-N键长分别为0.195 8nm,0.195 7nm,而Cu和N原子的共价半径和离子半径(括号内)分别为0.112(0.074)nm和0.074(0.171)nm,半径之和为0.186nm和0.245 nm,可见Cu-N键的共价键成分很大。四唑环骨架的平均键长(N-N, C-N)分别为0.135 8nm、0.136 4nm、0.135 3nm、0.135 4nm,处于孤立的N-N单、双键长分别为0.144 9nm和0.125 2nm,C-N单、双键长分别为0.147 1nm和0.127 3nm,说明四唑环具有很强的共轭性。
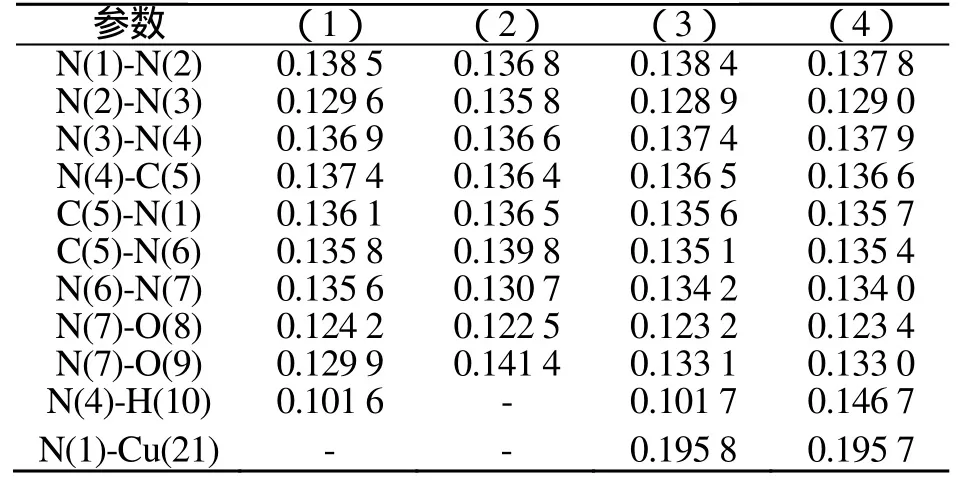
表1 BLYP/DNP优化几何键长 (nm)Tab.1 The optimized bond length by BLYP/DNP method
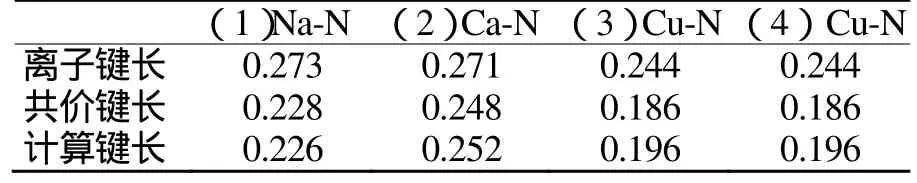
表2 理论键长与计算键长比较Tab.2 The bond length comparison of theory and calculation
2.1.2 键角
表3为BLYP/DNP优化几何键角,分析表3数据可知:硝氨四唑的四唑环骨架原子形成的键角在104~112°变化,大多数键角偏离120°(正常sp2杂化)而接近于正五边形的内角108°。这表明四唑环内存在一定张力,这是造成化合物不稳定的原因之一。
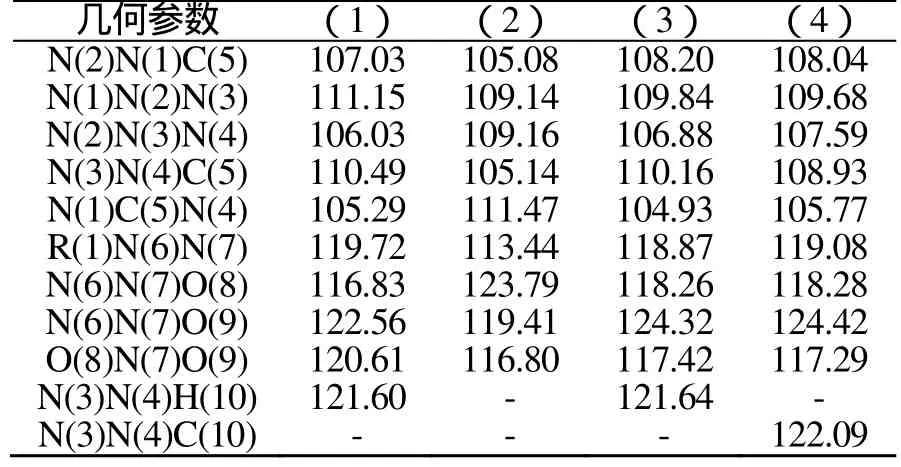
表3 BLYP/DNP优化几何键角 (°)Tab.3 The optimized bond angle by BLYP/DNP method
2.2 原子上净电荷
表4为BLYP/DNP水平下原子上的净电荷。由表4可见,中性分子中N(2)和N(3)原子上的电荷几近为零,仅(2)中N(2)、N(3)因与Ca相邻而具有较多的电荷。C(5)、N(7)和H原子均具正电荷,N(4)和N(l)原子上负电荷较多且均明显高于N(2)和N(3)。原子上负电荷越多,亲核性越强,其与金属成键形成配合物的能力亦愈强。因此,N(4)、N(l)是硝氨四唑化合物的主要配位中心。(3)、(4)由于结构对称,电荷分布也完全对称,因此,只讨论其半边分子即可。
硝氨基中O(8)和O(9)带有较多负电荷,且O(9)的负电荷均较O(8)多,这是由于各化合物中O(9)与金属原子距离较近。
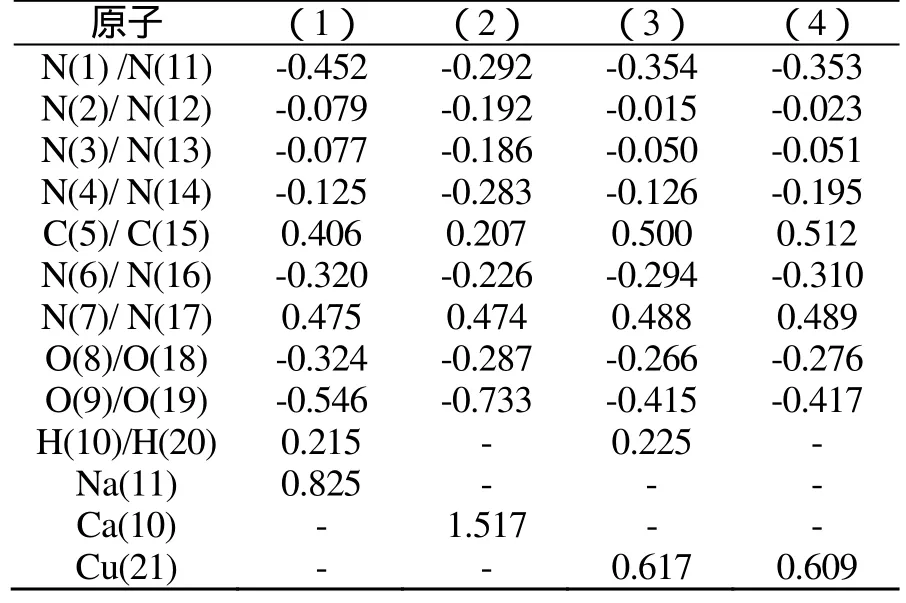
表4 BLYP/DNP水平下原子上的净电荷Tab.4 The atom charges by BLYP/DNP method
2.3 分子总能量、前线轨道能级及其差值
最高占有分子轨道(HOMO)是按照分子中的构造原理最后被占据的分子轨道,最低未占分子轨道(LUMO)则是紧接其后的能量较高的分子轨道。二者一起构成了分子的前线轨道。同类起爆药中,前线分子轨道能量与感度有着密切关系。前线轨道HOMO和LUMO及其附近的分子轨道对物质的活性(感度)影响很大,HOMO具有优先提供电子的作用,LUMO具有接受电子的重要作用。LUMO和HOMO的能级及其电子密度分布可分别反映该分子的亲电性、亲核性及亲电/亲核部位。LUMO能级在数值上与分子的电子亲和势相近,而HOMO能级在数值上近似于分子的第一电离能。能隙(△E)是LUMO轨道能级与HOMO轨道能级的能量差值,它反映了电子从占据分子轨道向空分子轨道跃迁的能力,一般而言,能隙越小,化学活性越大。认识起爆药分子的前线轨道及其分布有助于确定各种基团的活性部位,探索激发反应机理。
表5列出了各个化合物的分子总能量、前线分子轨道HOMO、LUMO的能量及其差值。分析可知:化合物分子总能量的绝对值增加,而前线分子轨道能隙逐渐减小,化合物稳定性降低。能隙△E分别为3.375 eV、2.951eV、0.865eV、0.969eV,预示(3)的活性最大,在一定条件下给予较少的能量就有可能发生化学反应。通过最易跃迁原理判据对4种5-硝氨基四唑金属盐的理论感度进行排序,从低到高依次为:Na NAT、CaNAT、Cu(1-mel-NAT)2、Cu(NAT)2,且CaNAT和Cu(1-mel-NAT)2的理论感度排序与撞击感度测试结果(CaNAT为5J,Cu(1-mel-NAT)2为>0.7J)[1-2]一致。

表5 分子总能量、前线轨道能级及能级差 (eV)Tab.5 The total energy, HOME energy, LUMO energy andenergy gap
2.4 红外光谱和热力学性质
红外(IR)光谱是化合物的基本属性,化合物的IR频率可用于计算其热力学性质。迄今为止,尚缺少硝氨四唑衍生物的IR和热力学性质的系统报道。图2为标题物的量子化学计算所得的红外波谱图。图3为标题物的BLYP/DND计算所得的热力学性质随温度的变化示意图。
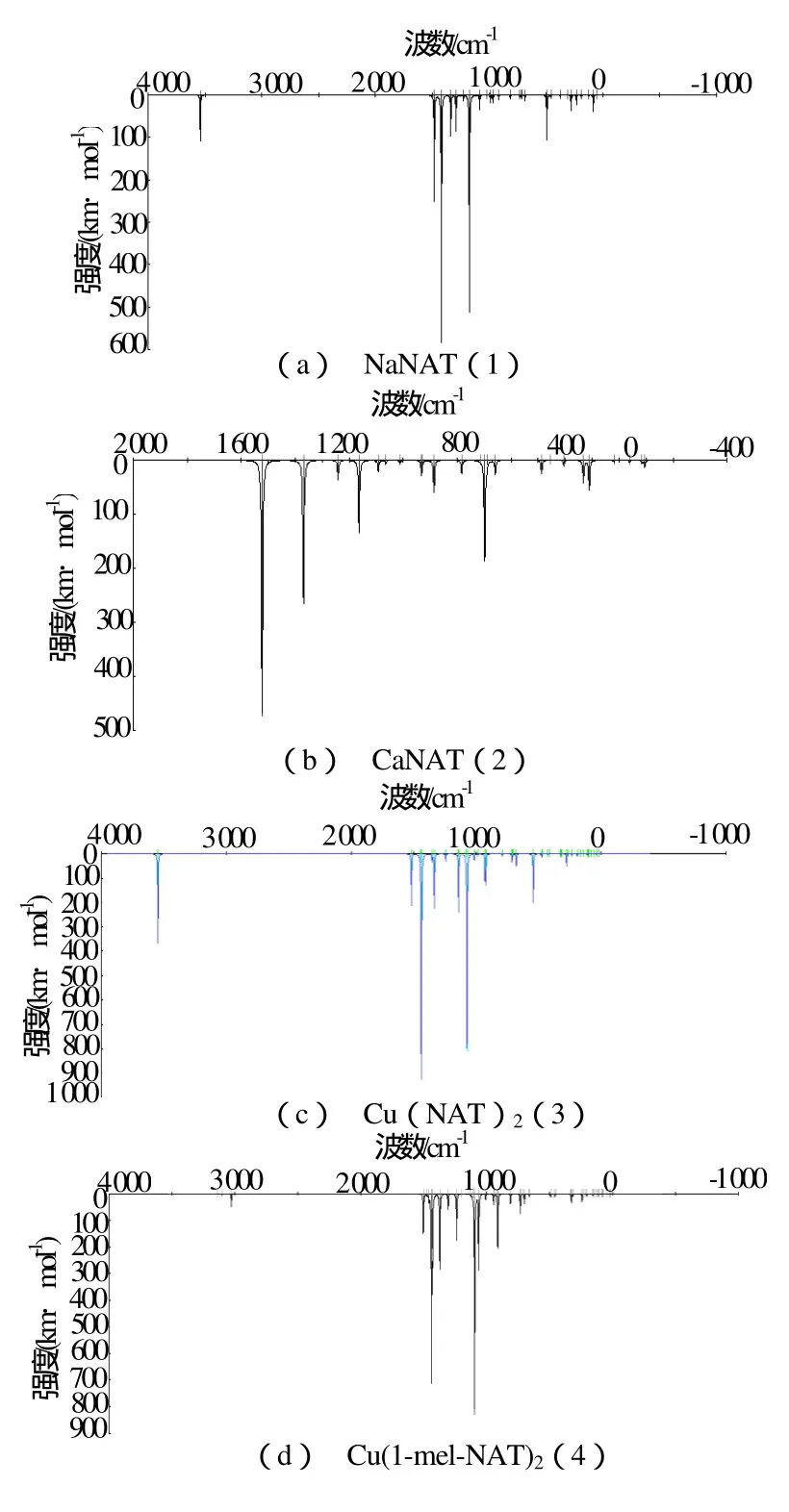
Fig.2 IR图 sp2e c tr标og题ra物m 的of IthRe光 ti谱tle图 compounds
由图2可见,硝氨四唑衍生物的IR谱具有4个特征区。在3 000cm-1以上,属于N-H键对称和反对称伸缩振动, (2)、(4)分子没有N-H键,故在此区域无吸收峰;其中两个很强的特征峰:1个在1 558.8~1 637.4 cm-1范围,对应于-NO2基中N=O键的不对称伸缩振动;另一特征峰在1 324.4~1 358.9 cm-1范围,对应于-NO2基中N=O键的对称伸缩振动;第4个特征区在小于1 250 cm-1波段,属于指纹区,可用来鉴别同分异构体,该区中较弱的峰主要由硝基的弯曲振动所引起。

图3 标题物的热力学性质——温度示意图Fig.3 The thermodynamics vs temperature of the title compounds
由图 3可见,除自由能外,标题物的各热力学函数均随温度升高而增加,归因于振动的贡献随温度升高而增大。标题物(1)和(2)的热力学性质相近,(3)和(4)的热力学性质相近,这是因为它们具有相似的几何结构和电子结构。
3 结论
通过对硝氨四唑金属化合物的量子化学计算,对计算所得的几何构型参数、电子结构、分子总能量、前线分子轨道能级及其差值等进行了分析。几何结构参数的分析表明:除5-硝氨四唑钙盐外,大多数原子处于同一个平面上,共面性比较高,结构较为稳定,具有电子离域性和分子共轭性。电子结构的分析表明:活性中心主要集中在金属离子和N(1)、N(4)上。分子总能量、前线分子轨道能级及其差值的分析表明:5-硝氨基铜盐的活性比较大即感度较高,在一定条件下给予较少的能量就有可能发生化学反应。
通过最易跃迁原理判据对4种5-硝氨基四唑金属盐的理论感度进行排序,从低到高依次为:NaNAT、CaNAT、Cu (1-mel- NAT)2、Cu(NAT)2。且CaNAT和Cu(1-mel-NAT)2的理论感度排序与撞击感度测试结果一致。
[1]Georg Geisberger,Klapötke,T.M.et al.Copper bis(1-methyl-5-nitriminotetrazolate): a promising new primary explosive[J].Eur.J.Inorg.Chem., 2007(30):4 743-4 750.
[2]Niko Fischer, T.M.Klapötke,et al.Calcium 5-nitrimi-notetrazolate—a green replacement for lead azide in priming charges[J].Journal of Energetic Materials, 2011(29): 61-74.
[3]T.M.Klapötke , Jörg Stierstorfer ,et al.New energetic materials: synthesis and characterization of copper 5-nitriminotetrazolates[J].Inorganica Chimica Acta, 2009(362):2 311-2 320.
[4]Thomas M.Klapötke,Hendrik Radies,et al.Alkali salts of 1-methyl-5-nitriminotetrazole –structures and properties[J].Z.Naturforsch,2007(62b):1 343-1 352.
猜你喜欢
四川大学学报(自然科学版)(2022年4期)2022-07-22
中学生数理化(高中版.高考理化)(2022年5期)2022-06-01
原子与分子物理学报(2022年3期)2022-03-05
中学生数理化(高中版.高考理化)(2021年5期)2021-07-16
火炸药学报(2020年5期)2020-10-27
青岛大学学报(工程技术版)(2019年2期)2019-09-10
兵器装备工程学报(2017年10期)2017-11-14
电子制作(2016年19期)2016-08-24
中学化学(2015年8期)2015-12-29
火炸药学报(2014年5期)2014-03-20
