
Alumina Supported Vanadium Oxide Catalysts for Residue Hydrotreating
2013-07-25 10:07JiaYanziLiDingjianyiYangQingheLiDadongSunShulingNieHong
中国炼油与石油化工 2013年1期
Jia Yanzi; Li Dingjianyi; Yang Qinghe; Li Dadong; Sun Shuling; Nie Hong
(Research Institute of Petroleum Processing, SINOPEC, Beijing 100083)
Alumina Supported Vanadium Oxide Catalysts for Residue Hydrotreating
Jia Yanzi; Li Dingjianyi; Yang Qinghe; Li Dadong; Sun Shuling; Nie Hong
(Research Institute of Petroleum Processing, SINOPEC, Beijing 100083)
In order to evaluate the role of vanadium in the hydrogenation (HYD) reaction, a series of alumina supported vanadium catalysts were prepared and characterized by SEM, XRD, Raman spectrometry,51V NMR, XPS, as well as TPR analyses. The catalytic performance of vanadium in HYD of model molecules (naphthalene) and real feedstock (Kuwait atmospheric residue) was studied after sulfidation of the catalysts. It can be concluded that the HYD capabilities of V/Al2O3catalysts are lower than that of conventional NiMo/Al2O3catalyst (RefNiMo). The V/Al2O3catalysts can only facilitate hydrogenation of the first ring of naphthalene, but have little effect on the further hydrogenation of tetralin. Owing to the different forms of metals and sulfur compounds in residue, the weak HYD activity of V/Al2O3catalysts is able to facilitate the HDM reaction of the residue, albeit with a slight effect on HDS activity.
residue; vanadium; hydrodemetallization; hydrodesulfurization
1 Introduction
Residue conversion to distillates by hydrogen addition technology has received more and more attention in recent years due to the increasing demand for clean fuels and the increasingly stringent environmental regulations. Among the different metals present in the heavy oils, Ni and V are the most important ones. After decomposition of the organo-metallic compounds, the metals are deposited on the catalyst surface. The metal deposits cause a loss of catalyst activity due to the covering of the active sites or pore plugging of the catalyst. However, the vanadium sulfide deposits during the hydrodemetallization (HDM) process can also act as catalyst for hydrotreating reactions[1-2]. In the late 1980s, Asaoka[3], Takeuchi[4], et al. claimed the autocatalytic activity of V3S4deposits for HDM reactions and firstly proposed its HDM mechanism.Ledoux, et al.[5]considered the activity of VxSy/Al2O3catalysts in thiophene hydrodesulfurizaiton (HDS). The catalytic activity was 30 times lower than that of a NiMo catalyst. In recent studies, Janssens[6]and Bonné[7]examined the HDM reactions of metalloporphyrins over sulfided V/ Al2O3catalysts, and found out that the HDM reactions of both nickel tetraphenylporphyrin (Ni-TPP) and vanadyl tetraphenylporphyrin (VO-TPP) are structure sensitive. It is well known that the residue consists of four hydrocarbon groups, i. e.: saturates, aromatics, resins and asphaltenes. Vanadium is mainly associated with porphyrin complexes which mostly exist in asphaltenes and resins. Because of the complexity of the residue, the activity of sulfided V/Al2O3catalysts and the impact of their structure on residue HDM and HDS are still not well elucidated.
The role of the vanadium deposits in the catalytic performance is of paramount importance since the catalyst can accumulate large amounts of vanadium during residue hydroprocessing[8]. The aim of this paper is to understand the relation between the structure and catalytic activity of alumina supported vanadium. The research in this paper will play a definite role in how to utilize the deposited vanadium and prepare new HDM catalysts with improved performance in residue hydrotreating.
2 Experimental
2.1 Catalyst preparation
The catalysts were prepared by pore volume impregnationof alumina support using a solution containing ammonium metavanadate. Since the solubility of ammonium metavanadate in water was limited, citric acid with the same molar weight was used as the solvent. The catalyst was dried at 120 ℃ for 4 h and then calcined at 400 ℃ for 3 h. The catalyst samples used in this study are listed in Table 1.
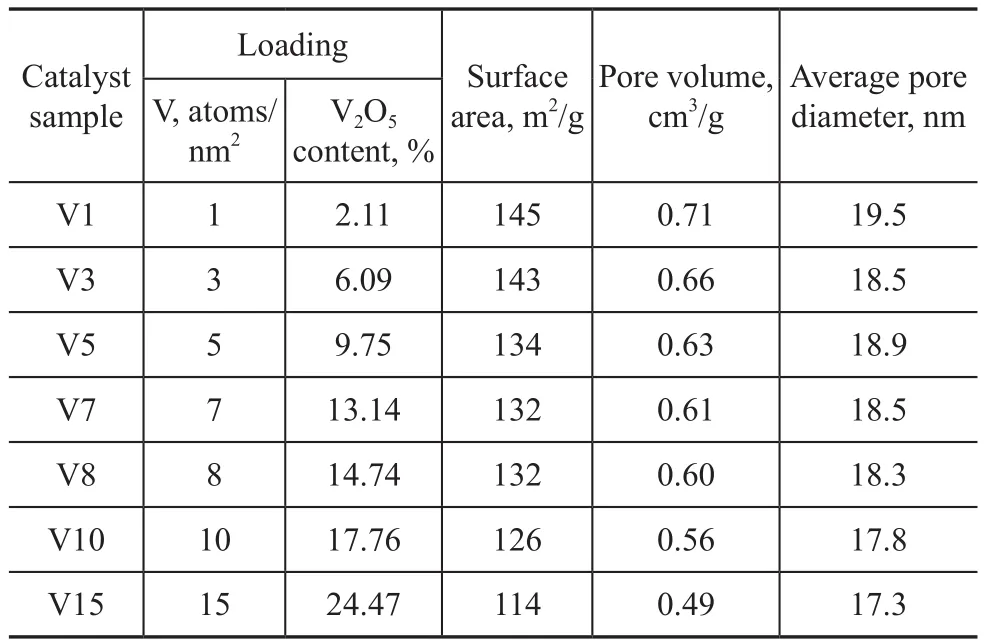
Table 1 Characteristics of alumina supported vanadium catalysts
A classical NiMo/Al2O3HDM catalyst designated as Ref-NiMo, containing 1.3% of NiO and 7.5% of MoO3on the same alumina support was used for comparison with the V/Al2O3catalysts.
2.2 Catalyst Characterization
The BET surface area and pore volume of catalyst samples were measured by nitrogen adsorption at -196 ℃ (using the Quantachrome AUTOSORB-6B surface area and pore size analyzer). The XRD patterns were collected on a Philips XPERT diffractometer equipped with a secondary graphite monochromator, operating at 40 kV and 30 mA and employing nickel-filtered Cu Kα radiation (λ=0.154 nm). The Raman spectra of the samples were obtained on a LabRAM HR confocal Raman spectrometer (Horiba JY). The solid state51V NMR spectra were recorded under static conditions at ambient temperature on a VARIANUNITYINOVA300M spectrometer, using a high-speed MAS Doty probe with zirconia rotors (6.0 mm in diameter). A pulse length of 2 μs and a recycle delay of 1 s were used for acquisition of the data. The cross sectional profile of vanadium in the catalysts was measured by a scanning electron microscope (SEM), model FEI Quanta 200F. The temperature-programmed reduction (TPR) experiments of the oxidic vanadium catalysts were performed on a Micromeritics AutoChem II2920 instrument in a 10% H2/Ar (by volume) gas stream at a flowrate of 50 mL/min; the furnace was heated up to 1000 ℃ at a rate of 10 ℃/min and a cold trap was equipped to condense the water vapor. The XPS spectra of the sulfided catalysts were acquired on a Thermo Fischer-VG ESCALAB 250 spectrometer. The C1s contamination peak at 284.8 eV was used for binding energy calibration. For sulfided catalysts, the sample preparation was carried out under controlled atmosphere (Ar with O2and H2O content being less than 15 μg/g). It should be noted that the above-used sulfided catalysts were freshly prepared according to the same ex-situ sulfidation procedures as used by the residue hydrotreating test in the following section.
2.3 Catalytic activity
2.3.1 Naphthalene hydrogenation
The naphthalene HYD activity tests were carried out in a continuous flow fixed-bed micro-reactor. The catalyst tests were carried out at 4.0 MPa and 300 ℃, with the catalyst loading equating to 1.0 g. A decane solution containing 1.0% naphthalene was used as the model feed, the experiments were conducted at a feed flow rate of 0.2 mL/min, and a H2flow rate of 200 mL/min. For each run, the weighed catalyst was diluted with quartz powder, and the solid mixture was placed in a stainless steel reactor (8.0 mm in inner diameter).
Prior to catalytic reaction tests, the catalysts were sulfided in situ with a mixture consisting of carbon disulfide (CS2, 5%) and cyclohexane (95%). The sulfidation was conducted at a total pressure of 4.0 MPa and a temperature of 360 ℃ for 3 h.
Under the experimental conditions, naphthalene was hydrogenated totrans-andcis-decalin via a partially hydrogenated intermediate, tetralin[9]. The liquid products of the hydrogenation reaction were analyzed by a gas chromatograph using a HP-1 column heated from 50 ℃ to 130 ℃and a flame ionization detector at 350 ℃.
2.3.2 Residue hydrotreating test
Residue hydrotreating experiments were carried out in a 500 mL batch reactor operated under the fixed-bed reaction conditions. The feed used was a Kuwait atmospheric residue (AR). The sulfur content of Kuwait AR was 5.0%,while the Ni and V content of Kuwait AR was 26.5 μg/g and 80.0 μg/g, respectively.
The experiments were carried out at a hydrogen pressure of 8.0 MPa, a reaction temperature of 380 ℃, and a reaction time of 2 h under a stirring rate of 200 r/min. A catalyst weight of 16.0 g was used to treat 250.0 g of feed. The catalyst was presulfided ex-situ for 10 h at a pressure of 3.2 MPa and a temperature of 320 ℃ with the sulfiding feed composed of 5% CS2in kerosene solvent.
Sulfur contents of the feed and effluents were measured by X-ray fluorescence analysis, while Ni and V contents were measured by Inductively Coupled Plasma Atomic Emission Spectrometry (ICP-AES).
3 Results and Discussion
3.1 SEM analysis
The cross sectional profile of the catalyst V15 was measured by SEM and presented in Figure 1. It showed that the vanadium pentoxide loaded on alumina deeply entered the catalyst pore cavity and was distributed unevenly. More vanadium atoms were concentrated on the periphery of catalyst particle. In earlier studies[10], it was also observed that V deposition formed a U-shape profile in aged catalysts obtained from commercial units. Since the molecular size of impreganation solution is smaller than vanadyl porphyrin in the residue, the U shape of the catalyst V15 was not obvious. But the homogeneous distribution profiles of other catalysts with lower vanadium content were observed (which is not shown here).

Figure 1 SEM cross sectional vanadium distribution of V15 catalysts
3.2 XRD analysis
Figure 2 shows the XRD patterns of V/Al2O3catalysts with different vanadium loadings. The characteristic diffraction peaks of vanadium pentoxide appeared at 2θof 15º, 20º, 26º, and 31º. It can be observed that there were weak peak humps of the catalyst V8 at around 15º and 26º. With a further increasing vanadium content, distinct peak humps at 2θof 15º, 20º, 26º and 31º appeared in the catalyst samples V10 and V15. It was also discovered that the average peak intensity of alumina decreased with an increasing vanadium loading.
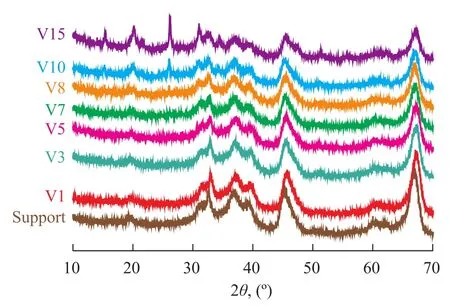
Figure 2 XRD patterns of V/Al2O3catalysts
3.3 Raman spectroscopy
Figure 3 shows the Raman spectra registered with the 325 nm exciting line. The high-surface area Al2O3support did not give rise to any Raman bands to verify vanadium oxide vibrations in the supported V/Al2O3catalysts. A significant change in catalyst structure was observed with the loading of vanadium pentoxide varying from 0 to 24.47%. The Raman spectrum of the 2.11% V/Al2O3sample (1 atom of V/nm2) exhibited a broad band at 1 008 cm-1arising from the terminal V=O vibration of the surface VO4species. The terminal V=O Raman band shifted from 1 008 cm-1to 1 025 cm-1with an increasing surface vanadium coverage[11]. This shift is related to distortions associated with polymerization of the surface VO4species as the vanadium oxide loading increases[12]. Except for the catalyst sample V1, samples with high surface vanadium loadings showed more pronounced bridging V-OAl vibration at 930 cm-1, indicating to the occurrence of polymeric vanadium species[13-14]. It can be seen from the XRD analysis that the crystalline V2O5species (with a Raman band at 995 cm-1) were not present up to 13.14% V in the alumina supported catalyst (with 7 atoms of V/ nm2) and only emerged at higher vanadium oxide surfacedensities. The appearance of crystalline V2O5in catalyst sample with 8 atoms of V/nm2reflected the completion of a surface vanadium monolayer on the Al2O3support[13,15].

Figure 3 Raman spectra of V/Al2O3catalysts
3.4 NMR spectroscopy
In order to get further information about the structure of the VOxcomplexes on the alumina support, static51V NMR spectra of catalyst samples were also recorded (Figure 4). Under the experimental conditions, the peaks of chemical shift between -500 and -600 were ascribed to polymeric tetrahedral coordinated vanadium species[16-17], while peaks of chemical shift between -270 and -320 were attributed to the octahedral coordinated ones[18]. The static spectrum of catalyst sample V1 showed a weak peak at -527, the characteristics of which allowed its assignment to tetrahedral vanadium species. This outcome was in good agreement with the previous Raman results. The static spectra of catalyst samples V3 and V5 consisted of a main signal with the maximum value appearing at -520 and shoulders occurring at approximately -270, denoting that the polymeric tetrahedral vanadium dominated on these two samples, whereas in catalyst samples V7 and V8, which had higher V coverage, the octahedral species were mostly detected[19]. The peaks at -1 250 in catalyst samples V10 and V15 indicated the appearance of square pyramidal crystallized vanadium pentoxide[17], which had been confirmed by XRD patterns. Besides, there were also octahedral coordinated vanadium species existing in catalyst samples V10 and V15 as verified by the peaks at -320. According to the XRD and Raman spectroscopic results, only tiny crystals were formed on the catalyst sample V8, which could explain why the peak at -1 250 relating to the crystallized vanadium pentoxide was not obvious in the NMR spectra.
Thus, a conclusion can be drawn out that at low surface concentration, the tetrahedral coordination of V5+is favored, but upon increasing the vanadium concentration, only octahedral coordinated species are mainly detected.

Figure 4 Static51V NMR spectra of V/Al2O3catalysts
3.5 TPR analysis
The results of TPR analysis of V/Al2O3catalysts are presented in Figure 5. It can be seen from Figure 5a that the pure alumina support showed a broad reduction peak with a maximum at 890 ℃, which could be attributed to impurities in the material. All V/Al2O3catalyst samples showed a reduction peak with a maximum at about 500 ℃. Figure 5b shows the relationship between temperature at the maximum reduction rate (tmax) and the vanadium loading of the investigated catalyst sample. As it can be seen from the curves that when the vanadium loading increased from 1 atom of V/nm2to 7 atoms of V/nm2;tmaxshifted to a lower value. When the vanadium loading exceeds 7 atoms of V/nm2, thetmaxshifted to higher values. It is argued that at low vanadium loading, tetrahedral coordinated vanadium was formed preferentially by the reaction of the precursor with surface hydroxyl groups of the support. With the increase of the vanadium loading, the small clusters could polymerize into large structures with a relatively weaker interaction with alumina than the small ones and became more prone to reduction. A further increase of the vanadium loading could lead to the formation of octahedral coordinated vanadium species, which would have an even weaker interaction with the support. And a further increase of vanadium loading must result in the formation of V2O5crystallites, which would be more difficult to be reduced[7].

Figure 5 TPR results: (a) TPR profiles of V/Al2O3catalyst samples; and (b) Observed temperature of the maximum reduction rate (tmax) from TPR vs. vanadium loading curve
3.6 Characterization of the sulfided catalysts
The V 2p3/2spectra for the sulfided solids (on the catalyst V5) are presented in Figure 6. The binding energy of V 2p3/2peak of the sulfided catalyst V5 was 517.3 eV, which was less than that of the oxidic catalyst V5. It is also noticed that the V 2p3/2spectrum was widened after the catalyst sulfidation. The decomposition of two vanadium species in a sulfur-rich environment was mainly evidenced by the existence of (V 2p3/2)4+(BE≈516.4 eV) and (V 2p3/2)5+(BE≈517.8 eV). This suggests that vanadium species exist in the VS2form under ex-situ sulfidation conditions.
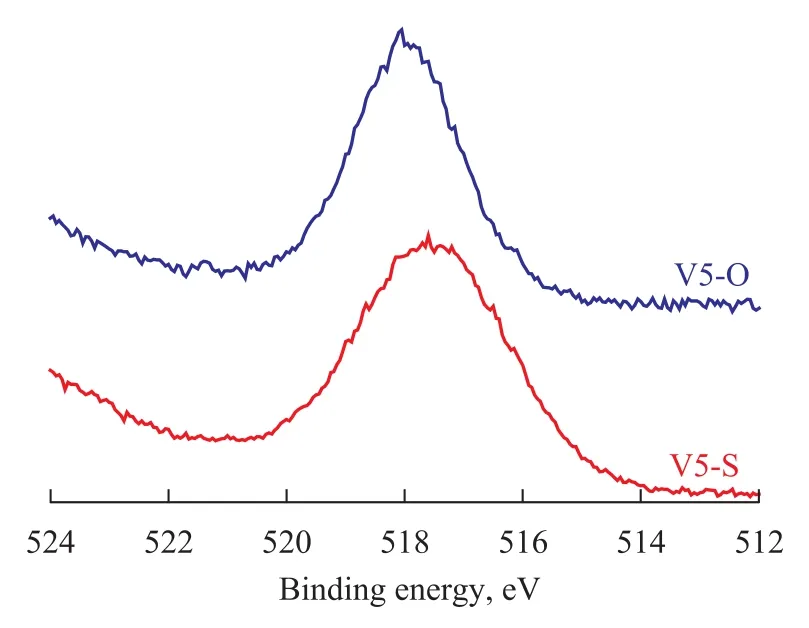
Figure 6 XPS V 2p3/2spectra of the catalyst V5 before and after sulfidation
3.7 Catalytic performance
3.7.1 Naphthalene hydrogenation
The HYD activities of the V/Al2O3and RefNiMo catalysts were measured upon treating the model feed. Table 2 shows the naphthalene hydrogenation activities of V/ Al2O3and RefNiMo catalysts.
The naphthalene conversion on the V/Al2O3catalysts increased with an increasing vanadium loading, while there was no decalin generated on the V/Al2O3catalysts. Based on the above characterization results, the impact of the vanadium loading of V/Al2O3catalysts on the naphthalene conversion can be summarized as follows. The relatively low hydrogenation activities of the catalyst V1 might be resulted from the presence of its active phase in tetrahedral coordination coupled with a strong interaction with the support. Below a monolayer coverage (<8 atoms of V/nm2), the increase in vanadium loading could make the small clusters expand to large structures to cause a weaker interaction with the support, as it can be inferred from the TPR results. The sulfur vacancies could be easily formed due to this weaker interaction, which was thought to be the active sites for hydrogenation[7].
What is worthy to be considered that above a monolayer coverage (>8 atoms of V/nm2), the naphthalene conversion on V/Al2O3catalysts increased slowly with the increase in vanadium loading. In Bonné’s studies[7], crystallites on the V/Al2O3catalysts had no hydrogenation capability. And through coverage of the active sites, crystallites can result in a reduction of the hydrogenation activity. According to the NMR spectra, except for V2O5crystallites, there are also octahedral coordinated vanadium species in catalysts samples V8, V10, and V15, which have weaker interaction with the alumina support and thereby higher hydrogenation activities than tetrahedral coordinated species. That is why the naphthalene conversion still increases with an increasing vanadium loading of the catalyst.
The activities of a RefNiMo catalyst were also measured. Its naphthalene conversation reached 100%, with the tetralin and decalin (includingtrans-andcis-decalin) yield equating to 12.62% and 87.38%, respectively. According to literature information[9,20], the hydrogenation of naphthalene proceeds via two steps: (i) the partial hydrogenation step (naphthalene to tetralin); and (ii) the complete hydrogenation step (tetralin totrans-andcis-decalin). It is learned that the rate of tetralin hydrogenation to decalin was one magnitude smaller than that of naphthalene hydrogenation to tetralin[21]. Table 2 shows that all catalyst samples had partial hydrogenation activity since they could only facilitate the conversion of relatively reactive naphthalene, but had little effect on the deep or complete hydrogenation of tetralin. So it seems that the reference catalyst (RefNiMo) showed higher catalytic activity in comparison with the V/Al2O3catalysts.

Table 2 Naphthalene hydrogenation activities of V/Al2O3and RefNiMo catalysts
3.7.2 Residue hydrotreating test
Hydrotreating of a Kuwait AR was also carried out on the V/Al2O3catalysts and the test results were compared with those achieved by the reference catalysts RefNiMo.
The rates for removal of nickel, vanadium and sulfur on V/Al2O3and RefNiMo catalysts are illustrated in Figure 7. It is observed that the vanadium removal rate on V/Al2O3catalysts increased with increase of the vanadium loading. The same trend was disclosed with respect to naphthalene conversion. The metalloporphyrins, which mostly exist in asphaltenes and resins, have been identified as the major metal-containing species in residues. Compared with the bimetallic NiMo catalyst (RefNiMo), the weak hydrogenation activity (for converting naphthalene to tetralin) of the V/Al2O3catalysts is able to facilitate the decomposition of asphaltenes and resins. Furthermore, the vanadyl porphyrin is demetallized through a reversible sequential mechanism via hydrogenated intermediates, and hydrogenation reaction is the rate determining step[6]. These two reasons may be related to the similar trend on conversion of naphthalene and vanadium species. In general, it is reasonable to conclude that with the enhancement of vanadium content, both the hydrogenation of naphthalene and the rate for removal of vanadium species are increasing, which can be correlated very well with the results of catalyst characterization.
With an increasing vanadium loading, the increase in nickel removal rate was identified as shown in Figure 7. Compared with the vanadium removal rate, the nickel removal rate was lower than that of vanadium on the same catalyst. It is well documented in the literature[22-23]that because the perpendicular oxygen atom linked to V atoms in vanadyl porphyrin is bonded strongly with the catalyst surface, V is easier to be removed than Ni from Niporphyrin which has no such oxygen links.

Figure 7 Rates for removal of nickel, vanadium and sulfur on V/Al2O3and RefNiMo catalysts
There was no appreciable change in sulfur removal rate of Kuwait AR with the increase of vanadium loading, and the sulfur removal rate remained at about 12% as shown in Figure 7. It is also noticed that with an increasing vanadium loading, the HDM activity of V/Al2O3catalysts could increase even up to the level of RefNiMo catalyst, whereas their HDS activity was much lower than that of RefNiMo catalyst. This phenomenon can be attributedto the different forms of sulfur and metals in heavy oils and the weak hydrogenation performance of V/Al2O3catalysts as evidenced by the data depicted in Table 2. Figure 8 shows the simulated asphaltene structural unit and the calculated bonding energies[24]. The reaction rate of HDM is faster than that of HDS due to lower bonding energy of V than that of S. On the other hand, the Ni-Mo-S phase has been found to have better hydrogenation performance than V-S phase judging from the above results. The difference between HDS and HDM activity of V/ Al2O3catalysts corroborates the conclusion that the weak hydrogenation activity of vanadium-based catalysts is able to facilitate the HDM reactions due to the relatively low bonding energy of the metal-coordinated bond. As regards the sulfur removal performance, the weak HYD activity of V/Al2O3catalysts can only facilitate the breakdown of the sulfur-containing molecules existing in the S—S bonds or C—S bonds having relatively low bonding energy. With respect to the C—S bonds having relatively higher bonding energy, the V/Al2O3catalysts do not seem to have much effect on their breakdown.
Therefore, with an increasing vanadium loading, a substantially higher HDM activity of V/Al2O3catalysts similar to that of RefNiMo catalyst was observed; while their HDS activities remained at a much lower level. This result is in agreement with Soogund and Guillard’s reports[25-26].
However, one should bear in mind that the residue hydrotreating reactions are exothermic, especially in terms of the HDS reactions. Higher removal rate of metals and lower removal rate of sulfur mean less heat release that could reduce the probability of hot spot formation in the catalyst bed during industrial runs. Besides that, the metal storage capacity is most important for the HDM catalysts. The satisfactory metal capacity needs a large pore volume and the absence of diffusion limitations that can allow for an efficient use of the catalyst. Consequently, it may be profitable to apply a vanadium based catalyst with a weak hydrogenation activity during the HDM process in order to avoid diffusion limitation, optimize the metal storage capacity, make use of the catalytic activity of the deposited vanadium, and reduce heat release.

Figure 8 Simulated results of asphaltene structural unit and calculated bonding energies (kJ/mol)
4 Conclusions
Characterization of alumina supported vanadium catalysts and their tests on hydrogenation of naphthalene showed that at lower vanadium loadings, the vanadium in tetrahedral coordination having a strong interaction with the support showed a relatively low hydrogenation activity. With an increasing vanadium content, the polymerization of the small clusters caused a weaker interaction with the support, resulting in a higher hydrogenation activity. A further increase of the vanadium content would form V2O5crystallites and octahedral coordination vanadium species with higher hydrogenation activities. Therefore the naphthalene conversion still increased at even higher vanadium loading.
The hydrogenation activity of the V/Al2O3catalysts waslower than that of the RefNiMo catalyst. The V/Al2O3catalysts could only facilitate the hydrogenation of the first ring of naphthalene, but showed little effect on further hydrogenation of tetralin.
Comparison of the V/Al2O3catalysts with RefNiMo catalyst conventionally used for residue HDM showed that with an increasing vanadium loading a substantially higher HDM activity similar to that of RefNiMo catalyst was observed, while their HDS activity was approximately two times lower than RefNiMo catalyst.
The vanadium appears as interesting new starting material for the preparation of residue HDM catalysts with high selectivity, viz. higher metals removal rate and lower sulfur removal rate than the conventional HDM catalysts, which also means less heat release, which would be possible to solve the problem of hot spots in the catalyst bed during industrial runs.
Acknowledgement:This work was supported by the National Basic Research Program of China (973 Program No. 2012CB224802).
[1] Sie S T. Catalyst deactivation by poisoning and pore plugging in petroleum processing[J]. Studies in Surface Science and Catalysis, 1980, 6: 545-569
[2] Devanneaux J, Gallez J P, Mariette L, et al. Kinetic comparison in demetallization by pure and active phase impregnated aluminas[J]. Preprints: Division of Petroleum Chemistry of American Chemical Society, 1985, 30: 84-95
[3] Asaoka S, Nakata S, Shiroto Y, et al. Characteristics of vanadium complexes in petroleum before and after hydrotreating[J]. ACS Symposium Series, 1987, 344: 275-289
[4] Takeuchi C, Asaoka S, Nakata S I, et al. Characteristics of residue hydrodemetallization catalysts[J]. Preprints: Petroleum Chemistry Division of American Chemical Society, 1985, 30: 96-107
[5] Ledoux M J, Michaux O, Hantzer S, et al. Hydrodesulfurization (HDS) poisoning by vanadium compounds: EPR and metal solid NMR analysis[J]. Journal of Catalysis, 1987, 106(2): 525-537
[6] Janssens J P, Langeveld A D, Moulijn J A. Characterisation of alumina- and silica-supported vanadium sulfide catalysts and their performance in hydrotreating reactions[J]. Applied Catalysis A: General, 1999, 179 (1/2): 229-239
[7] Bonné R L C, Steenderen P, Moulijn J A. Selectivity of γ-Al2O3supported vanadium catalysts in the hydrodemetallisation (HDM) of Ni-TPP and VO-TPP[J]. Preprints: Fuel Chemistry Division of American Chemical Society, 1991, 36: 1853-1863
[8] Hauser A, MarafiA, Almutairi A, et al. Comparative study of hydrodemetallization (HDM) catalyst aging by Boscan feed and Kuwait atmospheric residue[J]. Energy and Fuels, 2008, 22 (5): 2925-2932
[9] Sapre A V, Gates B C. Hydrogenation of aromatic hydrocarbons catalyzed by sulfided cobalt oxidemolybdenum oxide/alpha-aluminum oxide reactivities and reaction networks[J]. Industrial and Engineering Chemistry Process Design and Development, 1981, 20: 68-73
[10] Furimsky E, Massoth F E. Deactivation of hydroprocessing catalysts[J]. Catalysis Today, 1999, 52 (4): 381-495
[11] Kim T, Wachs I E. CH3OH oxidation over well-defined supported V2O5/Al2O3catalysts: Influence of vanadium oxide loading and surface vanadium-oxygen functionalities[J]. Journal of Catalysis, 2008, 255 (2): 197-205
[12] Tian H, Ross E I, Wachs I E. Quantitative determination of the speciation of surface vanadium oxides and their catalytic activity[J]. Journal of Physical Chemistry B, 2006, 110 (19): 9593-9600
[13] Weckhuysen B M, Keller D E. Chemistry, spectroscopy and the role of supported vanadium oxides in heterogeneous catalysis[J]. Catalysis Today, 2003, 78(1-4): 25-46
[14] Kanervo J M, Harlin M E, Krause A I, et al. Characterization of alumina-supported vanadium oxide catalysts by kinetic analysis of H2-TPR data[J]. Catalysis Today, 2003, 78 (1-4): 171-180
[15] Wachs I E. Raman and IR studies of surface metal oxide species on oxide supports: supported metal oxide catalysts[J]. Catalysis Today, 1996, 27 (3/4): 437-455
[16] Concepción P, Navarro M T, Blasco T, et al. Vanadium oxide supported on mesoporous Al2O3preparation, characterization and reactivity[J]. Catalysis Today, 2004, 96 (4): 179-186
[17] Blasco T, López Nieto J M. Nuclear magnetic resonance studies on supported vanadium oxide catalysts[J]. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 1996, 115: 187-193
[18] Steinfeldt N, Müller D, Berndt H. VOxspecies on aluminaat high vanadia loadings and calcination temperature and their role in the ODP reaction[J]. Applied catalysis A: General, 2004, 272 (1/2): 201-203
[19] Blasco T, Galli A, López Nieto J M, et al. Oxidative dehydrogenation of ethane andn-butane on VOx/Al2O3catalysts[J]. Journal of Catalysis, 1997, 169 (1): 203-211
[20] Montesinos-Castellanos A, Zepeda T A. High hydrogenation performance of the mesoporous NiMo/ Al(Ti, Zr)-HMS catalysts[J]. Microporous and Mesoporous Materials, 2008, 113 (1/2/3): 146-162
[21] Ito K, Kogasaka Y, Kurokawa H, et al. Preliminary study on mechanism of naphthalene hydrogenation to form declines via tetralin over Pt/TiO2[J]. Fuel Processing Technology, 2002, 79 (1): 77-80
[22] Ancheyta J, Maity S K, Betancourt G, et al. Comparison of different Ni-Mo/alumina catalysts on hydrodemetallization of Maya crude oil[J]. Applied Catalysis A: General, 2001, 216 (1/2): 195-208
[23] Chen Y W, Hsu W C. Hydrodemetallation of residue oil over CoMo/alumina-aluminum phosphate catalysts in a trickle bed reactor[J]. Industrial and Engineering Chemistry Research, 1997, 36 (7): 2526-2532
[24] Nie Hong, Yang Qinghe, Dai Lishun, et al. Development and commercial application of key technology for efficient conversion of heavy oil[J]. Petroleum Processing and Petrochemicals, 2012, 43(1): 1-6 (in Chinese)
[25] Soogund D, Lecour P, Daudin A, et al. New Mo-V based oxidic precursor for the hydrotreatment of residues[J]. Applied Catalysis B: Environmental, 2010, 98 (1/2): 39-48
[26] Loos M, Ascone I, Goulon-Ginet C, et al. X.A.F.S. study of model vanadium sulphide phases suspected to form on HDM catalyst surfaces[J]. Catalysis Today, 1990, 7 (4): 515-529
Recieved date: 2012-10-22; Accepted date: 2012-12-22.
Dr. Jia Yanzi, Telephone: +86-10-82368862; E-mail: jiayz.ripp@sinopec.com.

- 中国炼油与石油化工的其它文章
- Hydrocarbon Composition of Different VGO Feedstocks and Its Correlation with FCC Product Distribution
- Commercial Applications of Paraxylene Adsorbents RAX-2000A and RAX-3000
- Et3NHCl-AlCl3Ionic Liquids as Catalyst for Alkylation of Toluene with 2-Chloro-2-methylpropane
- Purification of Crude Glycerol from Waste Cooking Oil Based Biodiesel Production by Orthogonal Test Method
- Degradation of Nitrobenzene-Containing Wastewater with O3and H2O2by High Gravity Technology
- Highlights on Planned Grassroots Styrene Units and Expansion of Existing Styrene Units in China