
遗传性心律失常疾病相关离子通道病变的研究进展*
2013-09-14 06:46陈明颢胡峻岩
中国病理生理杂志 2013年3期
陈明颢, 胡峻岩
(1南方医科大学南方医院,广东 广州510515;2广州医学院第三附属医院急诊科,广东广州510150)
心源性猝死(sudden cardiac death,SCD)是常见的猝死病因。2006年北京阜外医院开展的流行病学调查研究表明,我国每年逾54.4万人死于SCD,而且发病率有逐年上升的趋势[1]。许多心源性猝死事件发生在心脏结构和冠状动脉正常的青年人中。这些患者无先兆症状和体征,多因为遗传缺陷产生的恶性心律失常致死。对于这类由于遗传因素导致心律失常的疾病,称为遗传性心律失常疾病(inherited arrhythmogenic diseases,IADs)[2]。致死性 IADs 主要包括长QT综合征(long QT syndrome,LQTS)、短QT综合征(short QT syndrome,SQTS)、Brugada综合征(Brugada syndrome,BrS)和儿茶酚胺能多形性室性心动过速(catecholaminergic polymorphic ventricular tachycardia,CPVT)等。
由于遗传性心律失常疾病的高死亡率及发病的不可预知性,人们一直都在研究其发病机制,寻求有效的预防和治疗方法。心律失常的病理生理机制是心肌细胞离子流的异常所致,而离子流由离子通道控制。1995年首次发现LQTS与离子通道基因突变直接相关,开创了心律失常遗传学研究的新时代[3]。近十几年以来,人们发现了越来越多遗传性离子通道病及其突变基因,明确了其发病机制,解释了许多不明原因的致死性心律失常。本文对几种离子通道病变在遗传性心律失常疾病发病机制中的作用进行综述。
1 几种遗传性心律失常疾病
1.1 LQTS LQTS的特征性表现是心电图QT间期延长以及心室复极波形态异常,心律失常为尖端扭转性室性心动过速(torsade de pointes,TdP)和心室颤动。LQTS是人们认识得最为全面的遗传性心律失常疾病,其有2种遗传形式。最常见的是常染色体显性遗传,这类患者仅表现为心脏的症状,称为Romano-Ward综合征(Romano-Wardsyndrome,RWS)。目前已发现位于13种基因上近千种的突变与RWS有关。常染色体隐形遗传型较罕见,约每百万人发生1.66例。这类患者除了QT延长和致死性心律失常外,还有感觉神经性耳聋症状,称为Jervell和Lange-Nielsen综合征(Jervell and Lange-Nielsen syndrome,JLNS)。JLNS分为 2种亚型:JLNS1和JLNS2型[4-5]。目前只有37个JLNS相关的基因突变的报道[6]。意大利学者的研究指出每2~3千个新生儿中即具有1个新生儿基因缺陷[7]。遗传性LQTS病死率高,未经治疗的患者10年病死率约为50%,是儿童和年轻人发作性晕厥和心源性猝死的主要原因[8]。
根据LQTS致病13种基因型的不同,将LQTS分为13型,见表1。LQTS的基因型已经成为LQTS危险程度分层的主要指标。基因型、性别和QT间期是LQTS的危险分层和预后的决定因素。根据基因型不同及其临床特性,ACC/AHA/ESC发布的2006年指南中,将 LQTS1、2、3 型进行危险度分层[9],以指导治疗,见表2。
基因型也是治疗方案的选择依据之一。例如,人们已经发现β受体阻滞剂对LQTS1、5、6、11型治疗效果较好,而对LQTS2型和LQTS3型效果较差;近期研究发现氟卡尼对于某些LQTS7型患者有效[10]。
1.2 SQTS Gussak 等[11]在 2000 年首先发现了一种QT间期缩短、并且伴有室性或房性心律失常以及猝死的遗传性疾病。此疾病为常染色体显性遗传,男女均可发病,猝死多见于年轻人,发病率不详。
目前已经发现 SQTS的6种基因型,见表1。SQTS的危险分层和治疗方法仍不明确。除了安置植入性心脏复律除颤器(implantable cardioverter defibrillator,ICD)治疗最有效外,奎尼丁和氟卡因也可减少恶性心律失常发生,但长期疗效尚不明确。奎尼丁对于SQTS1型效果较好,可作为安置ICD前的过渡治疗。
1.3 BrS BrS 由 Brugada等[12]于 1992 年首先报道,是一种编码离子通道基因异常所致的家族性原发性疾病,遗传方式为常染色体显性遗传伴不完全外显遗传,发病率约为5/100 000,亚洲人群发病率高。临床上主要病例为男性,目前尚不明确此病患者性别差异的原因。已发现8个基因与BrS有关,见表1。BrS的基因测定不能用于危险程度分层,主要用于筛查无症状患者。
1.4 CPVT CPVT是一种家族遗传性严重的心律失常疾病。CPVT平均发病年龄7~9岁,未经治疗的患者80%在40岁之前就发生室速、室颤、晕厥或猝死,死亡率30%。
目前发现 RyR2和CASQ2两个基因变异与CPVT有关,大约占总数的60%[13]。CPVT遗传学研究在分子水平上阐明了其产生心律失常的机制。动物实验证明延迟后除极是其发生机制[14]。更重要的是,CPVT遗传学研究有助于诊断及发现新的治疗方法。一项长期随访研究,基因检测的阳性率于确诊者约64%,有症状亲属89%,无症状亲属10%[15]。目前发现钠离子通道阻滞剂氟卡尼可能抑制DAD发生[16]。氟卡尼在CPVT中的应用正在临床试验中。
2 离子通道病变与遗传性心律失常
2.1 钾离子通道与遗传性心律失常 钾离子通道是目前发现最复杂的一类离子通道,分为延迟整流钾通道、瞬时外向钾通道、内向整流钾通道、三磷酸腺苷敏感钾通道和乙酰胆碱敏感性钾通道五类。心肌细胞钾通道决定心肌静息电位、心率及动作电位的形状和时程。
2.1.1 钾离子通道与LQTS 目前发现LQTS中有7个基因型与钾通道有关,分别为 LQTS1、LQTS2、LQTS5、LQTS6、LQTS7、LQTS11 和 LQTS13 型,其共同特点是相关基因突变导致通道功能缺失,使得钾离子从细胞内释放减少,导致复极时间延长。
缓慢延迟整流钾离子流(IKs)是心肌细胞复极过程中3相期的主要外向离子流之一,是对抗L型钙通道的内向离子流以终止平台期并最终完成复极的重要离子流。因此,LQTS1、LQTS5和LQTS11型相关的KCNQ1、KCNE1和AKAP9基因突变导致延迟整流钾通道功能受损,IKs被抑制,动作电位时程延长,即心电图上所表现的QT间期延长。钾离子通道的失活,心肌细胞的复极时间延长,使得原本正常的兴奋传导过程由于部分细胞不应期的延长而出现异常,为心律失常的出现提供了条件;后除极则是心律失常的诱发因素,后除极主要是由内向钙离子电流所致。
快速延迟整流钾离子流(IKr)是心肌细胞2、3相期主要复极电流之一,它在动作电位起始时很小,而在动作电位2、3相时明显增加。IKr增加,动作电位时程缩短;反之,IKr减弱,使心肌动作电位平台期延长和早期后除极,引起心律失常。LQTS2和LQTS6型中KCNE2和HERG基因突变导致IKr通道激活减慢、失活加快,通道开放缓慢,关闭迅速,使得钾离子外向复极电流减弱,复极化延长。
KCNJ2基因编码内向整流钾离子通道(IKl),调节IKl离子流,形成3期复极并维持静息膜电位。LQTS7型相关的KCNJ2基因突变,使得IKl减少,复极延长。
LQTS13型是通过一个携带KNCJ5基因突变的中国家族证实的。KNCJ5基因定位于11q23.3~24.3,编码的是乙酰胆碱依赖型钾通道Kir3.4的亚单位,研究表明其突变干扰了IKAch的功能[17],使复极时程延长。
2.1.2 钾离子通道与SQTS SQTS中有3种基因突变导致钾离子通道的异常,分别为KCNE2、KCNQ1和KCNJ2,与之相对应的分为 SQTS1、SQTS2和SQTS3型。这3种致病基因也与LQTS有关,只是突变所产生的钾离子通道功能不同。与LQTS突变抑制钾离子通道相反,SQTS中,3种基因的突变均为“获得功能”性突变,使钾离子流增加,复极时程缩短。
2.1.3 钾离子通道与BrS BrS中有2个基因型与钾离子通道有关。KCNE3基因编码电压门控钾离子通道KV4.3的β亚单位,可调节多种钾离子通道电流,如一过性外向离子流(Ito)和IKs等。Delpon等[18]通过SSCP法直接测序筛查105名BrS患者的离子通道基因,在4名病人KCNE3中均发现错义突变R99H,提示KCNE3与BrS有关。KCNE3基因突变为BrS6型。
HCN4基因突变为BrS8型。HCN4基因编码的HCN4蛋白参与构成超极化激活阳离子电流(If)通道结构[19],该基因是周期性核苷酸超极化电压依赖钾离子通道成员。该基因突变可致If减少。
2.2 钠离子通道与遗传性心律失常 钠离子通道是位于细胞质膜上的一种跨膜糖蛋白,通常由α亚基和β亚基组成,是心肌细胞膜去极化重要的阳离子通道之一。钠离子通道主要是选择性允许Na+跨膜通过,维持细胞兴奋性及其传导性。
2.2.1 钠离子通道与LQTS LQTS3型与SCN5A基因有关。SCN5A基因是电压门控性钠通道家族的一员,其编码快速内向钠离子流(INa)相关的钠通道(NaV1.5)的 α 亚单位[20]。SCN5A 基因突变使得NaV1.5增强,INa增加,导致动作电位延长。
CAV3基因与LQTS9型有关,其所编码的caveolin 3是参与细胞信号转导和胞内吞噬作用的一种蛋白。在心脏中,caveolin 3与钠离子通路蛋白相互作用。LQTS9型基因突变使钠通道延迟失活,钠离子流增加。
LQTS10型是SCN4B基因突变所致。SCN4B基因编码心脏钠离子通路的β4亚单位[21]。此基因突变使得钠通道延迟失活,钠离子流增加。
这些变异均造成钠离子通道“获得功能”性突变,使钠通道关闭延迟,钠离子在动作电位后期异常持续进入细胞内,持续进入的阳离子导致细胞恢复静息膜电位时间延长,所以动作电位时程延长。临床表现为QT间期延长。
LQTS12型与SNTA1基因突变有关。SNTA1基因定位于20q11.2,编码心脏 α1-互生蛋白(α1-syntrophin)。这种蛋白是通过大分子复合物连接细胞外基质和细胞内骨架一类蛋白[22-23]。SNTA1基因突变通过细胞膜的Ca2+-ATP酶(PMCA4b)释放了抑制物一氧化氮合酶,通过心脏钠通道亚硝基化导致钠离子流峰值增加和后移。
2.2.2 钠离子通道与BrS 目前发现BrS中有4个基因与钠离子通道有关。与LQTS3型的突变相反,SCN5A基因发生“丧失功能”性突变时,导致动作电位1相末期Na+内流减弱,瞬间外向钾离子流显著增强,使得动作电位平台期消失,导致心室跨室壁电压形成,在某些触发因素影响下产生2相折返,诱发多形性室速或室颤,患者表现为晕厥或猝死[24]。近期通过小鼠模型证明BrS发病机制既有心肌细胞除极的异常,又有复极的异常[25-27]。
BrS2型由GPD1-L基因突变所致。GPD1-L是编码甘油-3-磷酸脱氢酶1蛋白的基因。研究发现GPD1-L基因与 SCN5A基因没有直接关系,但是GPD1-L基因的一个杂合错义突变A280V会影响SCN5A基因的表面膜蛋白密度以及有功能通道的数量,因为其电生理学表现类型是SCN5A基因表达减少的结果,所以提示GPD1-L基因与BrS有关[28]。
SCN1B基因突变为BrS5型。SCN1B基因编码具有修饰功能的心肌钠离子通道的β1亚单位[29]。β1亚单位在心脏中的生物功能是增加钠离子内流来改变NaV1.5通道的功能;而SCN1B基因突变则会钝化或抑制该功能[29]。
SCN3B基因突变为 BrS7型。SCN3B也属于SCNB基因家族,编码心肌钠离子β3亚单位以传导钠离子内流。SCN3B一个突变基因与 SCN5A、SCN1B基因共同在TSA201细胞中表达时,发现它会影响 NaV1.5 通道功能,使得 INa内流减少[30]。
2.3 钙离子通道与遗传性心律失常 钙离子通道主要功能为调节细胞内Ca2+浓度,几乎存在于所有可兴奋细胞中。肌细胞钙通道可以分为电压门控型钙通道和钙释放通道两种。钙通道是一种重要的阳离子内流通道,其开放可以引起动作电位2相平台期形成及钙释放通道的激活。
2.3.1 钙离子通道与LQTS LQTS8型(Timothy综合征)是一种罕见的LQTS。患者表现为QT间期明显延长,伴有并趾、房室传导阻滞、先天性心脏病变、自闭症、神经发育障碍和免疫功能低下等其它症状[31]。LQTS8型由于CACNA1c基因功能获得性突变所致病。CACNA1c基因定位于12p13.3,编码电压门控钙通道的α亚单位,CACNA1c基因突变导致电压依赖钙通道失活功能丧失,引起细胞内钙超载及平台期产生持续去极化的钙离子流[32],使复极延迟。
2.3.2 钙离子通道与SQTS 已发现SQTS有3个基因型与钙离子通道有关。CACNA1c基因、CACNB2b基因和CACNA2D1基因分别编码L型钙离子通道的α1、β和α2δ1亚单位,其突变导致L型钙离子通道功能丧失,使内向钙离子流减少,动作电位时限缩短,QT 间期缩短[33-34]。
2.3.3 钙离子通道与BrS BrS3型由CACNA1c基因突变所致。CACNA1cf编码心脏钙离子通道的α1亚单位[34]。BrS3型为此基因功能丧失性突变,影响电压门控钙离子通道(CaV1.2),使得ICa内流减少。
CACNB2b编码心脏钙离子通道的β亚单位[34],其功能丧失性突变形成BrS4型。基因突变通过影响电压门控钙离子通道(CaV1.2),使得ICa内流减少是其发病机制。
编码钙离子通道的基因突变会产生既有BrS又有SQTS的混合型表现。研究发现CACNA2D1基因突变也与BrS有关[35],BrS与SQTS6型可能是等位基因突变所致,是否为新的基因型及两者关系还需进一步研究。
2.3.4 钙离子通道与CPVT RyR2基因位于1q42~43,表达RyR2(一种钙ryanodine受体)。RyR2通道主要分布在心肌细胞肌浆网上,调节胞浆内游离钙的浓度和平衡[36]。该基因突变,使得RyR2通道过度开放,导致舒张期肌浆网内Ca2+漏到细胞内,细胞胞浆内Ca2+浓度增加,诱发延迟后除极和触发活动增强,导致恶性心律失常。
CASQ2基因位于1p13.3~11,编码钙离子结合蛋白。Lahat等[37]在以色列7个家族中发现常染色体隐形遗传的CPVT,并在CASQ2基因的高度保守区发现D307H突变。D307H突变通过降低肌浆网腔中有效Ca2+浓度,细胞内钙调节失常,诱发快速性心律失常发生。
3 展望
总结上述离子通道疾病可以发现,几种离子通道病变都是相同的基因发生突变,但由于突变后蛋白表达功能的不同,导致了不同的病理表现,例如LQTS1型与SQTS2型、LQTS3型与BrS1型等。
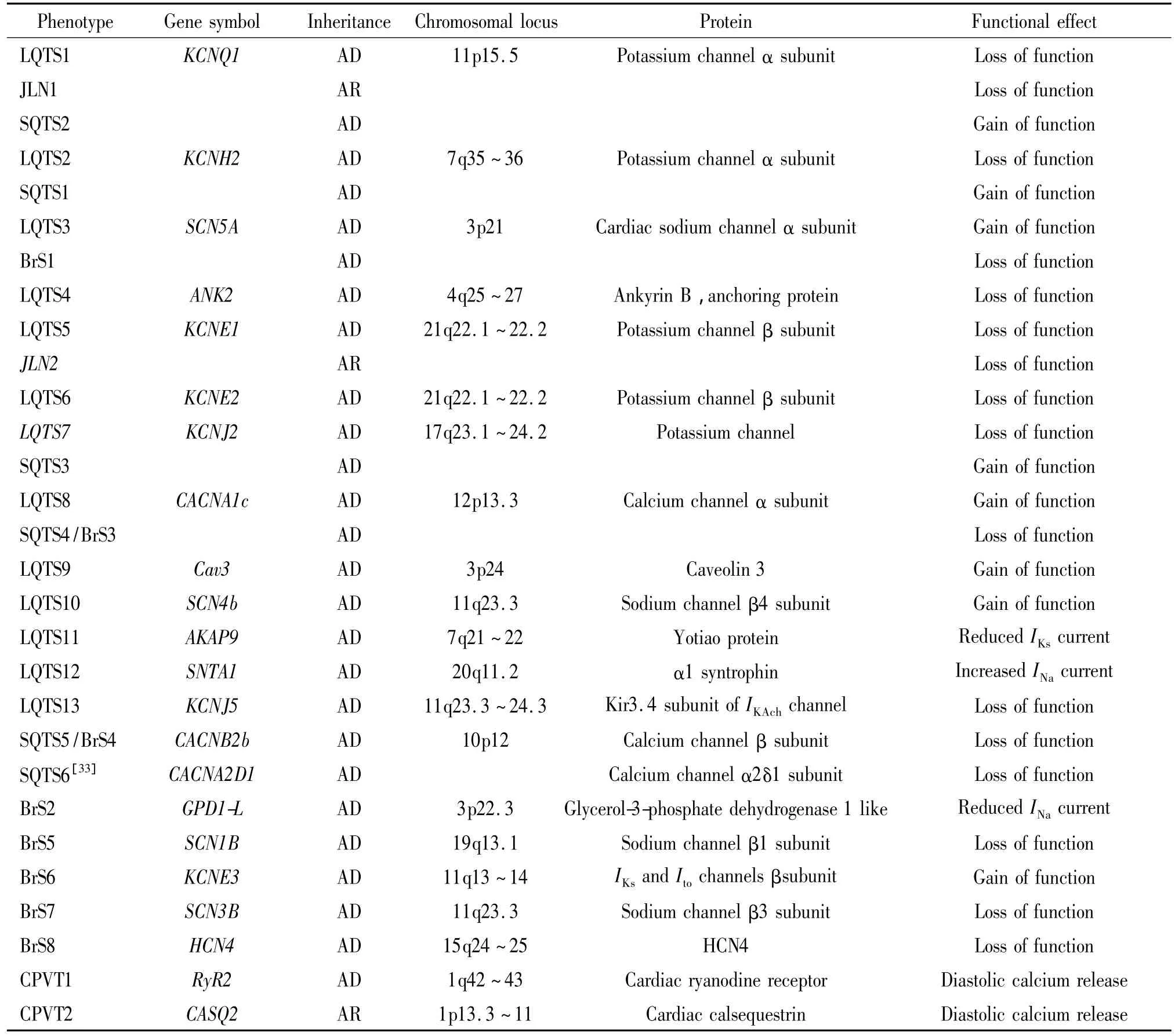
表1 遗传性心律失常疾病的基因位点和基因型Table 1.Genetic loci and genes associated with inherited arrhythmogenic diseases
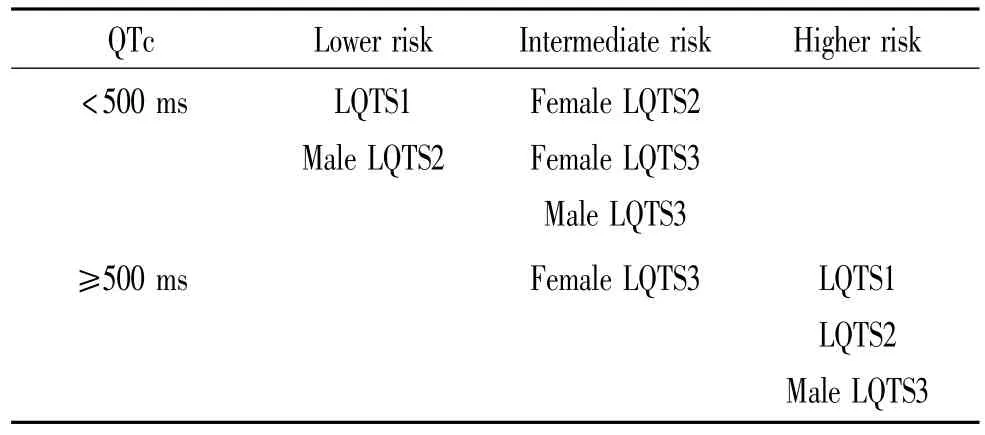
表2 LQTS患者危险度分层Table 2.Risk stratification in long QT syndrome
古人曰“上医治未病”,随着科技的发展将逐步实现这一想法。2011年美国心律学会/欧洲心律学会发布了《心脏离子通道病与心肌病基因检测专家共识》[38],怀疑IADs的患者可以通过基因分析,进行遗传学咨询、早期诊断、风险评估,提前应用药物或安装ICD干预,避免猝死的发生。遗传学研究更重要的意义在于根据患者的基因型,进行个体化治疗,并且使得基因治疗和通道选择性治疗成为可能。我国人口多,民族多,潜在的患者人数也很高,因此,进行深入的遗传学研究,明确国人基因突变的特点在预防和治疗心源性猝死中具有重要意义。
[1] Hua W,Zhang LF,Wu YF,et al.Incidence of sudden cardiac death in China:analysis of 4 regional populations[J].J Am Coll Cardiol,2009,54(12):1110-1118.
[2] Lehnart SE,Ackerman MJ,Benson DW Jr,et al.Inherited arrhythmias:a National Heart,Lung,and Blood Institute and Office of Rare Diseases workshop consensus report about the diagnosis,phenotyping,molecular mechanisms,and therapeutic approaches for primary cardiomyopathies of gene mutations affecting ion channel function[J].Circulation,2007,116(20):2325-2345.
[3] Keating MT.Molecular genetics of long QT syndrome[J].Soc Gen Physiol Ser,1995,50:53-60.
[4] Jervell A, Lange-Nielsen F. Congenitaldeaf-mutism,functional heart disease with prolongation of the Q-T interval,and sudden death[J].Am Heart J,1957,54(1):59-68.
[5] Schwartz PJ,Spazzolini C,Crotti L,et al.The Jervell and Lange-Nielsen syndrome:natural history,molecular basis,and clinical outcome [J].Circulation,2006,113(6):783-790.
[6] Gao Y,Li C,Liu W,et al.Genotype-phenotype analysis of three Chinese families with Jervell and Lange-Nielsen syndrome[J].J Cardiovasc Dis Res,2012,3(2):67-75.
[7] Arnestad M,Crotti L,Rognum TO,et al.Prevalence of long-QT syndrome gene variants in sudden infant death syndrome[J].Circulation,2007,115(3):361-367.
[8] Kaufman ES.Arrhythmic risk in congenital long QT syndrome[J].J Electrocardiol,2011,44(6):645-649.
[9] Zipes DP,Camm AJ,Borggrefe M,et al.ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death:a report of the American College of Cardiology/A-merican Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines(Writing Committee to Develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death)[J].J Am Coll Cardiol,2006,48(5):e247-e346.
[10] Barajas-Martinez H,Hu D,Ontiveros G,et al.Biophysical and molecular characterization of a novel de novo KCNJ2 mutation associated with Andersen-Tawil syndrome and catecholaminergic polymorphic ventricular tachycardia mimicry[J].Circ Cardiovasc Genet,2011,4(1):51-57.
[11] Gussak I,Brugada P,Brugada J,et al.Idiopathic short QT interval:a new clinical syndrome?[J].Cardiology,2000,94(2):99-102.
[12] Brugada P,Brugada J.Right bundle branch block,persistent ST segment elevation and sudden cardiac death:a distinct clinical and electrocardiographic syndrome.A multicenter report[J].J Am Coll Cardiol,1992,20(6):1391-1396.
[13] Hayashi M,Denjoy I,Extramiana F,et al.Incidence and risk factors of arrhythmic events in catecholaminergic polymorphic ventricular tachycardia[J].Circulation,2009,119(18):2426-2434.
[14] Kang G,Giovannone SF,Liu N,et al.Purkinje cells from RyR2 mutant mice are highly arrhythmogenic but responsive to targeted therapy[J].Circ Res,2010,107(4):512-519.
[15] Sy RW,Gollob MH,Klein GJ,et al.Arrhythmia characterization and long-term outcomes in catecholaminergic polymorphic ventricular tachycardia[J].Heart Rhythm,2011,8(6):864-871.
[16] Watanabe H,Chopra N,Laver D,et al.Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans[J].Nat Med,2009,15(4):380-383.
[17] Yang Y,Liang B,Liu J,et al.Identification of a Kir3.4 mutation in congenital long QT syndrome[J].Am J Hum Genet,2010,86(6):872-880.
[18] Delpon E,Cordeiro JM,Nunez L,et al.Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome [J].Circ Arrhythm Electrophysiol,2008,1(3):209-218.
[19] Ueda K,Hirano Y,Higashiuesato Y,et al.Role of HCN4 channel in preventing ventricular arrhythmia[J].J Hum Genet,2009,54(2):115-121.
[20] Wang Q,Shen J,Li Z,et al.Cardiac sodium channel mutations in patients with long QT syndrome,an inherited cardiac arrhythmia[J].Hum Mol Genet,1995,4(9):1603-1607.
[21] Medeiros-Domingo A,Kaku T,Tester DJ,et al.SCN4B-encoded sodium channel β4 subunit in congenital long-QT syndrome[J].Circulation,2007,116(2):134-142.
[22] Ueda K,Valdivia C,Medeiros-Domingo A,et al.Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex[J].Proc Natl Acad Sci U S A,2008,105(27):9355-9360.
[23] Cheng J,Van Norstrand DW,Medeiros-Domingo A,et al.α1-syntrophin mutations identified in sudden infant death syndrome cause an increase in late cardiac sodium current[J].Circ Arrhythm Electrophysiol,2009,2(6):667-676.
[24] Antzelevitch C.The Brugada syndrome:diagnostic criteria and cellular mechanisms[J].Eur Heart J,2001,22(5):356-363.
[25] Matthews GD,Martin CA,Grace AA,et al.Regional variations in action potential alternans in isolated murine Scn5a+/-hearts during dynamic pacing[J].Acta Physiol(Oxf),2010,200(2):129-146.
[26] Martin CA,Zhang Y,Grace AA,et al.Increased right ventricular repolarization gradients promote arrhythmogenesis in a murine model of Brugada syndrome[J].J Cardiovasc Electrophysiol,2010,21(10):1153-1159.
[27] Martin CA,Guzadhur L,Grace AA,et al.Mapping of reentrant spontaneous polymorphic ventricular tachycardia in a Scn5a+/-mouse model[J].Am J Physiol Heart Circ Physiol,2011,300(5):H1853-H1862.
[28] London B,Michalec M,Mehdi H,et al.Mutation in glycerol-3-phosphate dehydrogenase 1 like gene(GPD1-L)decreases cardiac Na+current and causes inherited arrhythmias[J].Circulation,2007,116(20):2260-2268.
[29] Watanabe H,Koopmann TT,Le Scouarnec S,et al.Sodium channel β1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans[J].J Clin Invest,2008,118(6):2260-2268.
[30] Hu D,Barajas-Martinez H,Burashnikov E,et al.A mutation in the β3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype[J].Circ Cardiovasc Genet,2009,2(3):270-278.
[31] Splawski I,Timothy KW,Sharpe LM,et al.CaV1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism[J].Cell,2004,119(1):19-31.
[32] Splawski I,Timothy KW,Decher N,et al.Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations[J].Proc Natl Acad Sci U S A,2005,102(23):8089-8096;discussion 8086-8088.
[33] Templin C,Ghadri JR,Rougier JS,et al.Identification of a novel loss-of-function calcium channel gene mutation in short QT syndrome(SQTS6)[J].Eur Heart J,2011,32(9):1077-1088.
[34] Antzelevitch C,Pollevick GD,Cordeiro JM,et al.Lossof-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation,short QT intervals,and sudden cardiac death[J].Circulation,2007,115(4):442-449.
[35] Burashnikov E,Pfeiffer R,Barajas-Martinez H,et al.Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death[J].Heart Rhythm,2010,7(12):1872-1882.
[36] Laitinen PJ,Brown KM,Piippo K,et al.Mutations of the cardiac ryanodine receptor(RyR2)gene in familial polymorphic ventricular tachycardia[J].Circulation,2001,103(4):485-490.
[37] Lahat H,Pras E,Olender T,et al.A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel[J].Am J Hum Genet,2001,69(6):1378-1384.
[38] Ackerman MJ,Priori SG,Willems S,et al.HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies:this document was developed as a partnership between the Heart Rhythm Society(HRS)and the European Heart Rhythm Association(EHRA)[J].Europace,2011,13(8):1077-1109.
猜你喜欢
——从一道浙江选考生物学试题谈起
中学生物学(2022年9期)2022-11-11
中国自行车(2022年5期)2022-09-06
昆虫学报(2021年7期)2021-08-18
宇航计测技术(2021年1期)2021-08-17
江苏农业科学(2021年1期)2021-03-15
心肺血管病杂志(2020年5期)2021-01-14
黑龙江工业学院学报(综合版)(2020年8期)2020-10-23
绿色科技(2017年10期)2017-07-05
湖北农业科学(2016年20期)2017-02-15
电子制作(2016年24期)2016-04-18
