
合成5-(2-溴丁酰基)-8-羟基喹诺酮的工艺改进
2013-11-19 11:08蒲凌翔张义文
合成化学 2013年6期
蒲凌翔, 肖 蓉, 张义文, 宋 航
(四川大学 化学工程学院,四川 成都 610065)
5-(2-溴丁酰基)-8-羟基喹诺酮(5, Chart 1)是有机合成中的重要中间体[1],可在其活性部位引入新的手性基团。1-氢-2-羰基-5-(L-亮氨酸甲酯)-8-羟基-喹诺酮(Ⅰ, Chart 1)是一类π-酸,π-碱型新型芳香族化合物,能够在几种不同类型的手性固定相上得到很好的拆分[2]。根据互为相反作用原理,用Ⅰ制备成手性固定相具有良好的拆分能力。

Chart1
.5是合成Ⅰ的重要中间体。如何高效、经济的合成5对Ⅰ的合成与应用具有积极的作用。
本文以8-羟基喹啉(1)为原料,经氧化反应制得8-羟基喹啉-1-氧化物(2); 2经乙酰化反应制得8-乙酰氧基喹诺酮(3); 3用K2CO3水解得8-羟基喹诺酮(4); 4经傅克酰基化反应合成了5(Scheme 1),其结构经1H NMR,13C NMR和IR确证。
本文对合成2~5的工艺进行改进。用改进方法合成的5,总收率从16%[1]提高至49%,反应时间从33 h[1]缩短至16 h。5的合成,对Ⅰ的高效合成提供了重要基础,为进一步开发和应用创造了条件。

Scheme1
1 实验部分
1.1 仪器与试剂
XRC-1型显微熔点仪(温度未校正);LC-20AT型高效液相色谱仪;SPD-M20A型二极管阵列紫外检测仪;Bruker AV 600 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);Nicolet 6700型红外光谱仪(KBr压片)。
所用试剂和溶剂均为分析纯,未经纯化直接使用。
1.2 合成
(1) 2的合成
在反应瓶中依次加入二氯甲烷60 mL和1 11.6 g(80 mmol),搅拌使其溶解;于30 ℃以下滴加20%过氧乙酸(AcO2H)48.0 mL(145 mmol),滴毕(约30 min),于30 ℃反应3 h(TLC跟踪)。加饱和硫代硫酸钠溶液除去未反应的AcO2H。分液,水层用二氯甲烷(3×25 mL)萃取,合并有机层,依次用饱和碳酸氢钠溶液(2×25 mL)、饱和食盐水(25 mL)及蒸馏水(25 mL)洗涤(以淀粉-KI试纸检测过氧乙酸是否完全除尽)。低温减压旋蒸回收二氯甲烷得棕黄色固体2 8.70 g,收率67.5%(28.0%[1])
(2) 3的合成
在反应瓶中依次加入2 3.22 g(20 mmol)和乙酸酐20.0 mL(200 mmol),搅拌下于100 ℃反应5 h。冷却至室温,于70 ℃以下旋干溶剂,剩余物用无水乙醇(3×20 mL)洗涤,过滤,滤饼干燥得米黄色固体3 3.70 g,收率91.1%(77.0%[1])。
(3) 4的合成
在反应瓶中依次加入3 4.06 g(20 mmol),碳酸钾2.76 g(20 mmol),蒸馏水5.0 mL及甲醇35.0 mL,搅拌下于室温反应1 h(4 h[1])。旋干溶剂,滴加10%盐酸至无气泡产生为止。过滤,滤饼用水洗涤,干燥得灰白色固体4 3.01 g,收率93.2%(84.0%[1])。
(4) 5的合成
在反应瓶中依次加入三氯甲烷50 mL, 4 3.22 g(20 mmol), 3-溴丁酰氯9.27 g(50 mmol)和无水三氯化铝8.01 g(60 mmol),搅拌下于40 ℃反应3 h。缓慢倾入冰水中,分液,水层用三氯甲烷(3×50 mL)萃取,合并有机层,旋蒸除溶,残余物用冰甲醇洗涤后,过滤,滤饼干燥得白色固体5 2.64 g, m.p.214 ℃~220 ℃,收率85.2%;1H NMRδ: 7.85~7.87(d,J=5.3 Hz, 1H, 1-H), 6.60~7.50(s, 4H, ArH), 10.95~10.96(d,J=2.5 Hz, 1H, 8-H), 4.86~4.90(t,J=4.0 Hz, 1H, 2′-H), 2.17~2.38(q,J=4.5 Hz, 2H, 3′-H), 1.11~1.24(t,J=4.5 Hz, 3H, 4′-H);13C NMRδ: 162.24(C2), 128.33(C3), 144.22(C4), 119.29(C5), 124.85(C6), 116.92(C7), 144.53(C8), 127.36(C9), 123.29(C10), 189.87(C1′), 54.19(C2′), 27.05(C3′), 11.96(C4′); IRν: 3 422, 3 113, 2 972, 2 888, 2 847, 1 760, 1 655,1 604, 1 404, 1 371, 1 341, 1 191, 837, 500~700(C-Br) cm-1。
2 结果与讨论
2.1 2的反应条件优化
(1) 氧化剂及用量
文献[1]方法用过氧化氢为氧化剂,收率仅28%。本文对文献[1]方法进行改进,用20%AcO2H代替30%过氧化氢作为氧化剂。
1 80 mmol, AcO2H为氧化剂,其余反应条件同1.2(1),考察AcO2H用量对反应的影响,结果见表1。由表1可见,氧化剂用量低于145 mmol时,Ac2OH受热分解,1反应不完全;高于145 mmol时,后处理不仅会多消耗硫代硫酸钠及饱和碳酸氢钠水溶液,收率也未见增加,说明用量已过量。AcO2H最佳用量为145 mmol(收率65.2%)。
(2) 反应温度
1 80 mmol, AcO2H 145 mmol,其余反应条件同1.2(1),考察反应温度对反应的影响,结果见表2。由表2可见,0 ℃时反应体系呈胶状,无收率;随着温度的升高,产率逐渐升高,30 ℃达最大值(65.3%); 40 ℃收率有所下降,可能是由于AcO2H受热分解过快所致。最佳反应温度30 ℃。
综上所述,合成2的最佳反应条件为:1 80 mmol, AcO2H 145 mmol,于30 ℃反应3 h,收率67.5%。该条件的优化,不仅使收率从文献[1]方法的28.0%提高至67.5%,而且降低反应温度至室温,避免了高温下过氧酸易爆的危险,降低了能耗,并采用一次性加料的方式简化了投料过程。

表1 催化剂用量对反应的影响*Table 1 Effect of catalyst amount on the reaction
*1 80 mmol,其余反应条件同1.2(1)

表2 反应温度对反应的影响*Table 2 Effect of temperature on the reaction
*1 80 mmol, AcO2H 1.8 eq,其余同表1
2.2 3的反应条件优化
由于文献[1]方法的反应温度较高(140 ℃),为此对其进行优化,以便找到更佳的反应温度。
2 20 mmol,其余反应条件同1.2(2),考察反应温度对反应的影响,结果见图1。
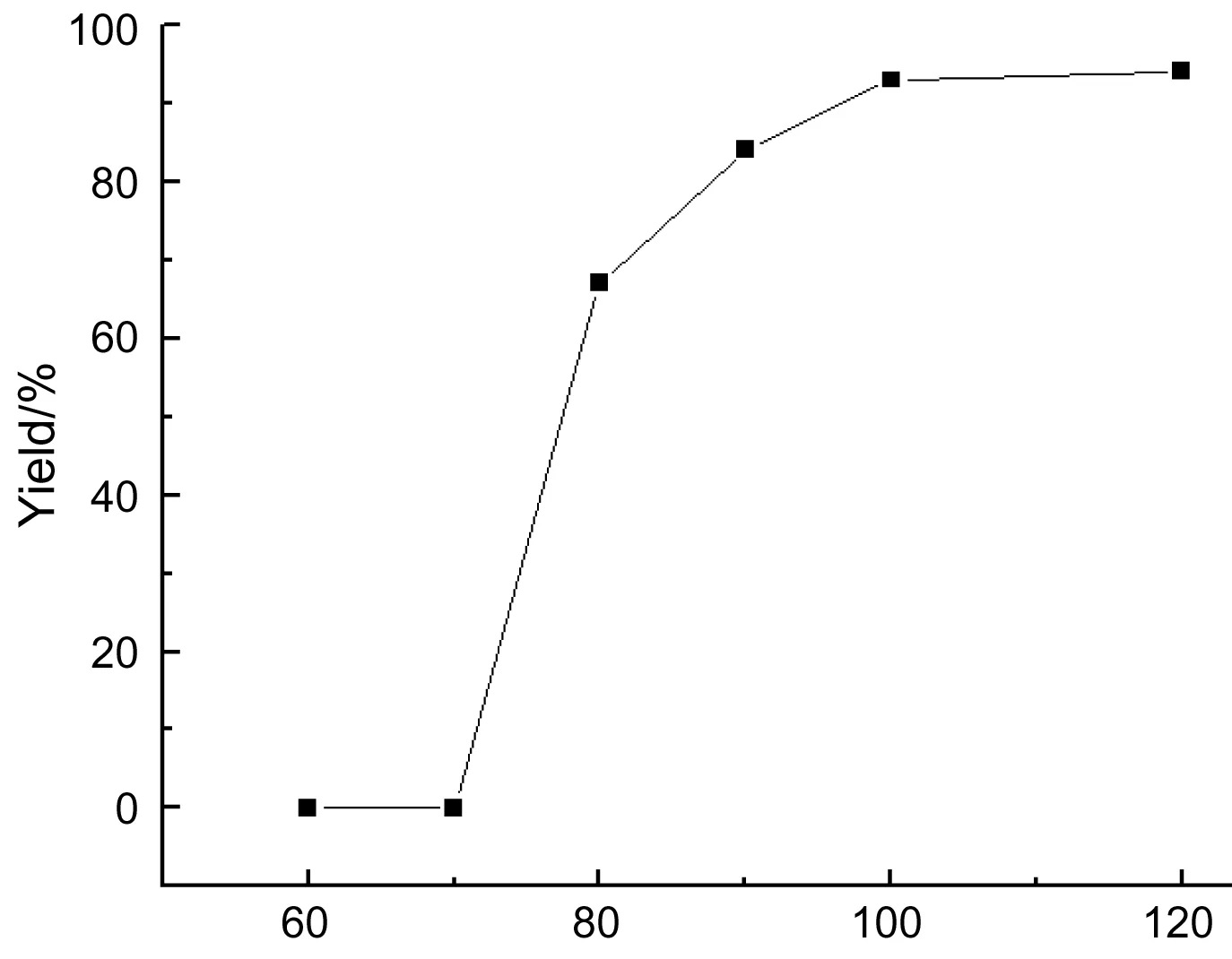
Temperature/℃图1 反应温度对反应的影响*Figure 1 Effect of temperature on the reaction*2 20 mmol,其余反应条件同1.2(2)
由图1可见,60 ℃时反应12 h, TLC检测未发现有3生成;在100 ℃时,几乎达到定量产率;温度继续升高产率未见增加。最佳反应温度100 ℃。
2 20 mmol,反应温度100 ℃,其余反应条件同1.2(2),考察反应时间对反应的影响。实验结果表明,最佳反应时间为5 h。
综上所述,合成3的最佳反应条件为:2 20 mmol,于100 ℃反应5 h。此工艺的改进,不仅较文献[1]方法降低了反应温度,而且收率从文献[1]方法的77.0%提高至91.1%。
2.3 4的反应条件优化
在4的合成中,文献[1]方法用浓盐酸作催化剂,浓盐酸在高温腐蚀性较强。本文参考文献[3]方法,采用K2CO3碱水解的方式制备4{由于未能找到良好的展开剂体系,实验采用高效液相色谱[HPLC,流动相:V(甲醇) ∶V(水)=3 ∶2;流速:0.8 mL·min-1;柱温:298.15 K;色谱柱:C18柱;紫外检测波长:254 nm]进行检测}。实验结果发现,于室温反应60 min后反应结束。此工艺的改进,避免了文献[1]方法中高温长时间回流的苛刻要求,降低了能耗及设备成本。用K2CO3替代盐酸作催化剂,加速了水解速度,使反应时间由4 h缩短至60 min,反应温度由80 ℃降至25 ℃。
2.4 5的反应条件优化
(1) 溶剂
按文献[1]方法使用CS2作溶剂合成5时发现,反应为非均相反应,影响传质,产物形成致密膏状物粘附于瓶壁,冷却后变硬,用冰水破坏产物络合物结构时很费时,收率较低。为此进行工艺改进,用CHCl3替代CS2作溶剂,反应体系呈均相,后处理操作容易。但以CHCl3为溶剂处理时会产生一种灰色的膏状物,即残留的3-溴丁酸, 可用甲醇洗除。在加入少量甲醇时,可以明显感觉到放热。而用甲醇洗的次数越多,产品的纯度就越低,推测放热可能破坏产物结构。文献[4]也提及到产物热不稳定的现象。而采用冰甲醇代替常温甲醇进行洗涤时,洗涤后的产物与常温甲醇洗涤后的产物的HPLC[流动相:V(正己烷) ∶V(异丙醇)=90 ∶10;流速:1.0 mL·min-1;柱温:298.15 K;色谱柱:DMB柱;紫外检测波长:254 nm]图对比发现,用冰甲醇替代普通甲醇进行洗涤,可显著提高产物的纯度,这也验证了文献[4]方法的稳定性差的说法,用冰甲醇洗涤可以维持它的稳定性。
(2) 反应温度和反应时间
4 20 mmol,其余反应条件同1.2(4),考察反应温度[25 ℃, 40 ℃, 50 ℃(文献[1]温度)及65 ℃(氯仿回流温度)]对反应的影响{以HPLC[流动相:V(正己烷) ∶V(异丙醇)=9 ∶1;流速:1.0 mL·min-1;柱温:298.15 K;色谱柱:DMB柱;紫外检测波长:254 nm]检测产物纯度作标准}。实验结果表明,25 ℃时基本不反应;50 ℃和65 ℃反应均有副产物生成;40 ℃以下反应产率高、副反应少。
考察反应时间(3 h, 5 h, 7 h)对反应的影响,从HPLC谱图(略)可见,反应3 h时产品纯度比较高,而5 h时已经产生了很多不同的杂质,7 h后样品的色谱图和5 h样品的色谱图一致。
综上所述,合成5的最佳反应条件为:4 20 mmol,以CHCl3为溶剂,于40 ℃反应3 h,收率85.2%。
改进工艺将CS2替换为氯仿,实现了均相反应,避免了原溶剂的易燃危险,后处理亦明显简化,收率由65%[1]提高至85.2%,反应时间由13 h缩短至3 h。
[1] 焦淑清,于莲,侯巍. 盐酸丙卡特罗合成工艺改进[J].中华医学写作杂志,2002,9(17):1380-1381.
[2] 唐琴,陈先勇,宋航. 丙卡特罗在三种刷型手性固定相上的直接拆分[J].分析测试学报,2010,29(4):407-410.
[3] S Chauhan, B Kalra, P Mohapatra. Oxidation of 1-naphthol and related phenols with hydrogen peroxide and potassium superoxide catalysed by 5,10,15,20-tetraaryl-porphyrinatoiron(Ⅲ)chlorides in different reaction conditions[J].Journal of Molecular Catalysis A:Chemical,1999,137:85-92.
[4] S Yoshizaki, K Tanimura, S Tamada,etal. Sympathomimetic amines having a carbostyril nucleus[J].Journal of Medicinal Chemistry,1976,19(9):1138-1142.
猜你喜欢
能源化工(2021年2期)2021-12-30
世界最新医学信息文摘(2021年12期)2021-06-09
科学与财富(2021年33期)2021-05-10
汽车零部件(2018年5期)2018-06-13
国外医药(抗生素分册)(2016年5期)2016-07-12
国外医药(抗生素分册)(2016年2期)2016-07-12
广东石油化工学院学报(2016年6期)2016-05-17
中国卫生标准管理(2015年25期)2016-01-14
合成化学(2015年5期)2015-03-26
石油化工应用(2014年2期)2014-03-11
