
遗传性结直肠癌的体系结构和诊治进展
2013-11-27 01:20丁振顾国利
中华结直肠疾病电子杂志 2013年4期
丁振 顾国利
遗传性结直肠癌约占全部结直肠癌的三分之一[1-2],依据是否继发于结肠息肉病,遗传性结直肠癌可分为遗传性非息肉病性结直肠癌(hereditary nonpolyposis colorectal cancer,HNPCC)和遗传性结肠息肉病(hereditary colorectal polyposis)两大类[3]。而遗传性结肠息肉病又依据病理类型的不同可分为腺瘤性息肉病综合征和错构瘤息肉病综合征两类,包括家族性腺瘤性息肉病(familial adenomatous polyposis,FAP)、基 因 相 关 性 息 肉 病 (MYH-associated polyposis,MAP)、遗传性色素沉着-消化系息肉病综合征(Peutz-Jeghers syndrome,PJS)、家族性幼年性结肠息肉病(familial juvenile polyposis coli,FJPC)、PTEN基因突变与常染色体显性遗传性错构瘤综合征(PTEN hamartoma tumor syndrome,PHTS)、遗传性混合息肉病综合征(hereditary mixed polyposis syndrome,HMPS)等一系列疾病(图1)。由于遗传病因特殊、病理特点突出,遗传性结直肠癌是目前临床肿瘤学研究的热点[4]。笔者对遗传性结直肠癌的体系结构和诊治进展加以系统总结,以加深临床医生对遗传性结直肠癌的认识,提高对上述疾病的诊治能力。
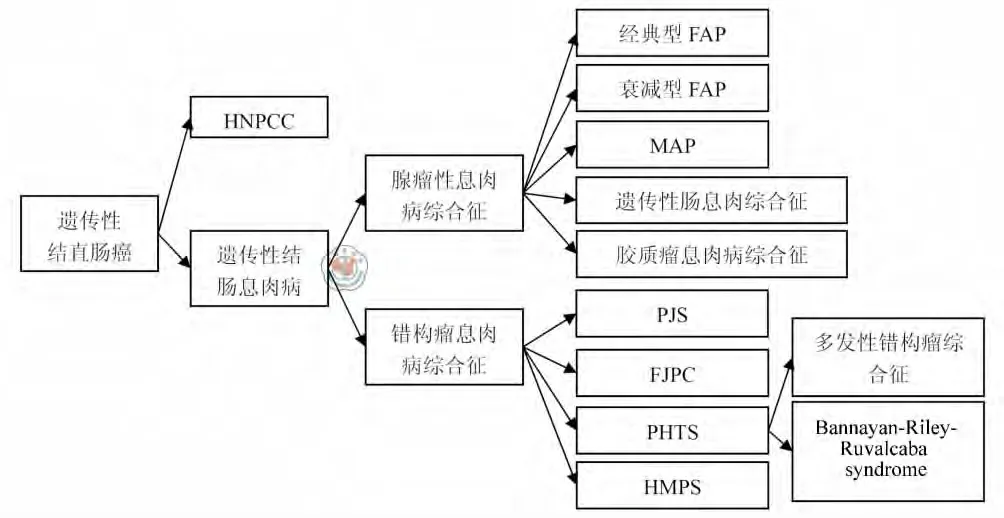
图1 遗传性结直肠癌的结构体系图
一、HNPCC
HNPCC又称林奇(Lynch)综合征,是一种由错配修复基因(mismatch repair gene,MMR)种系突变而引起的常染色体显性遗传病[4]。约占全部大肠癌的5% ~15%。
(一)遗传学基础
目前已证实,MMR的种系突变以及由突变而引起的微卫星序列不稳定(microsatellite instability,MSI)是HNPCC发生的遗传学基础[5]。HNPCC患者遗传获得胚系突变的MMR基因,相应编码蛋白的缺失将影响DNA错配修复功能,使细胞具有了恶变的可能性。而DNA复制错误的增加将使基因组DNA的微卫星序列产生不稳定性—即MSI。
(二)临床病理特点
HNPCC临床病理特点非常突出。目前国内外对其临床病理特征形成以下共识[5-10]:(1)发病年龄早,中位年龄约为44岁;(2)近段结肠多见,约70%位于脾曲近侧;(3)多原发大肠癌明显增多;(4)肠外恶性肿瘤发生率高;(5)低分化腺癌、粘液腺癌常见,且伴有淋巴细胞浸润或淋巴样细胞的聚集;(6)垂直遗传和家族聚集;(7)多呈膨胀性生长,而非浸润性生长。预后较好。
(三)临床诊治现状
1、临床诊断标准:目前HNPCC的临床诊断沿用HNPCC国际合作组织于 1998年制定的 Amsterdam 标准Ⅱ[4,11-12]:(1)家族中3例以上患有组织学证实的HNPCC相关肿瘤(包括大肠癌、子宫内膜癌、小肠癌、肾盂输或尿管癌),其中1例为另2例的一级亲属;(2)累及连续的两代人;(3)至少1例发病年龄小于50岁。
全国遗传性大肠癌协作组2003年制定了中国人HNPCC家系筛检标准[13]:家系中至少有2例组织病理学明确诊断的大肠癌患者,其中的2例为父母与子女或同胞兄弟姐妹的关系,并且符合以下一条:(1)至少1例为多发性大肠癌患者(包括腺瘤);(2)至少1例大肠癌发病早于50岁;(3)家系中至少1人患HNPCC相关肠外恶性肿瘤(包括胃癌、子宫内膜癌、小肠癌、输尿管或肾盂癌、卵巢癌、肝胆系统癌)。同时,还制定了实验室筛检策略:HNPCC可疑家系均应进行hMLH1、hMSH2免疫组化和MSI检测。两项均阴性者无需进行突变检测分析;两项之一阳性者,则需进行 hMLH1和hMSH2基因种系突变检测分析。
2、临床治疗现状:手术仍是目前治疗HNPCC的主要方式,但是临床上对具体手术方式仍有争论。有学者认为[14]:HNPCC患者在首次确诊时就应施行全结肠切除术,以避免多原发结直肠癌发生的风险,并且降低了对HNPCC患者残留结肠漏诊的几率。然而,另有学者认为[15]:HNPCC肿瘤谱广泛,即使切除了全部结直肠,其它器官也可能患HNPCC相关肿瘤。目前,NCCN指南对HNPCC初发大肠癌患者仅是建议考虑行全结肠或次全结肠切除术。因此,对于初发HNPCC患者应综合考虑其临床分期、预后、随访条件及个人意愿,向患者提出预防性手术的建议,在患者知情同意的前提下才考虑行预防性手术切除治疗。
二、FAP
FAP是腺瘤性息肉综合征的代表,发病率约为1/7000~22000。又可分为经典型(classical familial adenomatous polyposis,CFAP)、衰减型 (attenuated familial adenomatous polyposis,AFAP)、MYH基因相关性息肉病(MYH-associated polyposis,MAP)、Gardner 综合征 (Gardner syndrome,GS)、Turcot综合征(Turcot syndrome,TS)等亚型[16]。
(一)经典型FAP
1、遗传学基础:FAP是由APC基因突变引起的常染色体显性遗传病[16-18]。APC基因突变导致其编码的APC蛋白功能性缺失,这可导致β-catenin在细胞质内的异常积聚并进入细胞核,从而启动和调节Wnt/β-catenin信号通路的下游靶基因(包括 c-myc、cyclinD1、MMP-7 和 ITF-2)的表达[9-10]。
2、临床病理特点[16-19]:(1)息肉数目多(大于 100个),多分布于左半结肠,其次为右半结肠,再次为横结肠;(2)息肉发生年龄早平均15岁,恶变年龄早(平均39岁),恶变率高,且多灶性恶变、转移早、预后差。如不治疗,患者多在45岁之前死于大肠癌;(3)以管状腺瘤、绒毛状腺瘤和管状绒毛腺瘤多见。直径一般小于1 cm,多数是宽基底,大于2 cm的腺瘤通常有蒂;(4)可伴发结肠外表现,如:胃息肉、十二指肠息肉、硬纤维瘤、先天性视网膜色素上皮增生等。
(二)衰减型FAP
1.遗传学基础:只是由于APC基因突变位点的不同,衰减型FAP显示出明显不同的临床表现[20-21]。
2.临床病理特点[20-22]:(1)息肉数目少(通常为10~100枚),且呈右半结肠分布趋势;(2)息肉发生晚(平均34岁)、恶变晚(平均57岁)、恶变率稍低(60%),如不治疗,患者死于大肠癌时间晚(平均59岁);(3)息肉多呈扁平状。除CFAP常见病理类型外,AFAP还可表现为一种特殊病理类型—锯齿状腺瘤;(4)常伴胃及十二指肠腺瘤(50% ~66%),伴发硬纤维瘤较少(10%),其余结肠外特征报道偶见,尚未见到伴发先天性视网膜色素上皮增生的报道。
3.临床诊断标准:目前尚无国际通用的AFAP诊断标准。有学者提出[23]:同一家系中至少有两个患者,结直肠腺瘤数目为10~99枚,诊断时年龄大于30岁,或家系中至少有一个患者结直肠腺瘤数为10~99枚,诊断时年龄大于30岁且其一级亲属中有伴发少量腺瘤的结直肠癌患者,同时家族成员中没有一例30岁之前腺瘤总数超过100枚者。
(三)MYH基因相关性息肉病
1.遗传学基础:MAP是由MYH基因突变而引起的常染色体隐性遗传病[24]。MYH基因在不同人种的MAP中具有不同类型的突变(无义、错义、框移、剪切位点的突变及产生截短蛋白)[25]。
2.临床病理特点:(1)息肉数目少(一般小于100个),左半结肠多发,其次为右半结肠。(2)息肉发生晚、恶变晚(平均46岁)、恶变率高(65岁前恶变率为100%)。(3)可伴有结肠外表现,如十二指肠息肉、先天性视网膜色素上皮肥大、乳腺癌、胃癌、骨肉瘤、甲状腺癌等,但较少出现[25-27]。
3.临床诊断标准:MAP确诊主要依赖于基因检测。临床上对于无显性遗传家族史,但息肉数目多于10个,或具有一些相关肠外表现的患者,即应考虑MAP;对MYH基因则应进行MYH全基因测序。对MAP患者的亲属应进行预防性基因检测。
(四)Gardner综合征(GS)
1.遗传学基础:绝大多数学者认为GS是FAP的一个特殊的临床亚型;其遗传学基础仍是APC基因的突变(多为密码子1403和1578的截短突变)[28-30]。
2.临床病理特点:(1)结直肠息肉数量多(大于100个),分布广泛。(2)胃和十二指肠息肉多见,但小肠息肉少见。(3)息肉生长多年后常在青壮年发病,且恶变率高。(4)骨瘤合并牙齿畸形和软组织肿瘤(皮脂腺囊肿、硬纤维瘤、脂肪瘤等)为其合并症,并可伴随其它瘤变(如甲状腺瘤、肾上腺瘤及肾上腺癌等)[28-31]。
3.临床诊断:具备结直肠多发息肉、骨瘤和软组织肿瘤这3大特征者即可确诊为GS。但临床上尚有一些不典型患者,可仅表现出结直肠息肉病而无肠外病变,或仅有肠外病变而无结直肠息肉病。GS的肠外病变通常多为潜在性存在,需针对性地仔细诊察才能发现。另外,如不重视家族史的调查也易造成漏诊。
(五)Turcot综合征
又称胶质瘤息肉病综合征(glioma-polyposis syndrome,GPS),是 FAP 的一种罕见亚型[32]。
1.遗传学基础:以往把TS发生完全归于APC基因的突变[29,32],但近年来发现:MMR基因在 TS中也扮演着非常重要的角色。甚至有学者建议将 TS也划归 HNPCC的范畴[32-35]。
2.临床病理特点:(1)发病率低,临床上非常罕见;(2)发病早(平均17岁),预后不良,多在发病数年内死于脑肿瘤;(3)结肠腺瘤性息肉数目多(100个左右),体积较大,全结肠分布,癌变率高且年龄较轻(20岁前恶变率100%);(4)神经胶质瘤多发于大脑的连个半球,少数出现在小脑、脑干及脊髓。其病理组织形态多种多样,如:成胶质细胞瘤、成神经管细胞瘤、星形细胞瘤、多形性成胶质细胞瘤等;(5)可有结肠外伴随病变,如胃十二指肠、小肠肿瘤、脂肪瘤、甲状腺癌、卵巢囊肿,皮肤咖啡牛乳色斑等。
(六)FAP临床治疗现状
目前临床上对于FAP的结直肠息肉仍主要采取外科手术治疗。手术方式有3类:(1)全结肠直肠切除+永久性回肠造口术:适用于全结直肠广泛息肉,并有距肛门6 cm内的直肠癌患者。该术式并发症多、患者生活质量差,现已很少应用;(2)全结肠直肠切除+回肠贮袋成形+直肠鞘内肛管吻合术:适用于结直肠内息肉无癌变、括约肌完好的50岁以下的患者。该手术盆腔感染率高,功能恢复不理想;(3)结肠全切除+直肠黏膜剥除+回肠贮袋肛管吻合术:因其切除全部大肠黏膜,彻底消除腺瘤再发与癌变的可能性,又不作永久性回肠造口,所以得以普遍应用。但手术复杂,并发症较多[36-37]。
三、错构瘤息肉病综合征
错构瘤指在发育中出现错误而形成的肿瘤[38]。既往认为错构瘤极少恶变,但现在研究发现,错构瘤的恶变比率较高[39]。PJS是错构瘤息肉病综合征的主要代表。
(一)遗传性色素沉着消化道息肉病综合征
又称黑斑息肉病,发病率约为1/25000,以皮肤黏膜色素斑、胃肠道错构瘤息肉和家族遗传性为三大临床特征[40-41]。
1.遗传学基础:LKB1/STK11编码一种丝氨酸/苏氨酸激酶。LKB1/STK11基因的突变使STK11蛋白激酶功能发生异常。通常,LKB1/STK11基因的胚系突变仅可在一部分患者中被检测出(60%家族性和50%散发性)。因此,有学者认为:LKB1/STK11基因突变位点和形式的多样性不仅与遗传背景有关,也可能与PJS患者生存的环境有关[40-45]。
2.临床病理特点:(1)息肉数目多,大小不一,全消化道分布,好发于空肠上段;(2)息肉可引起急慢性腹痛、肠套叠、肠扭转、肠梗阻及胃肠道出血等并发症;(3)约有60%患者有明确或可疑家族史,部分可出现隔代遗传,真正散发性PJS非常罕见;(4)随着患者年龄的增长,息肉恶变的风险随之增加;(5)可伴发肠外肿瘤,如乳腺癌、女性生殖系统肿瘤、睾丸支持细胞瘤及神经结神经胶质瘤等[40,45]。
3.临床诊治现状:(1)诊断标准:消化道多发错构瘤性息肉伴皮肤、黏膜色素沉着。被诊断为PJS患者应进行LKB1/STK11和FHIT基因的检测[13];(2)鉴别诊断:临床上PJS需与Cronkhite-Canada综合征相鉴别。Cronkhite-Canada综合征发病晚,是一种获得性、非遗传性疾病,此病可能与感染、缺乏生长因子及砷中毒有关。虽也可表现为消化道息肉和黏膜色素沉着,但其还有脱发、指(趾)甲萎缩脱落的特征性临床表现;(3)治疗:目前,手术配合内镜治疗是PJS的主要治疗方式[46],但分子靶向治疗则将是 PJS治疗的方向[47]。双气囊电子小肠镜对于PJS的诊断和治疗具有非常大的优势[48-49]。手术主要是针对由息肉引起的肠梗阻、套叠及出血等并发症进行治疗[46]。
(二)家族性幼年性结肠息肉病(FJPC)
FJPC以结直肠多发幼年性息肉为特征[39,50],发病率约为1/100000,“幼年性”指的是肉的形态,而非发病年龄。
1.遗传学基础:BMPR1A和SMAD4基因突变是FJPC的遗传学病因。BMPR1A和SMAD4基因突变生成的无功能产物可造成TGF-β/SMAD信号通路中SMAD蛋白复合物失活,从而影响其下游基因的表达,导致肿瘤形成[39,50-52]。
2.临床病理特点:FJPS可以分为3型:(1)婴儿型:较少见,多在出生后数周内出现粘液性腹泻、呕吐、便血等症状,从而继发贫血和营养不良;也可出现肠梗阻、直肠脱垂和肠套叠。如不手术,常死于消化道出血、肠梗阻及腹泻引起的营养不良;(2)结肠型:最常见,息肉数目多在50~200个,多位于乙状结肠和直肠。以便血、粘液便及结肠息肉脱垂为主要症状。发病年龄较早(平均6岁),恶变率较高;(3)胃肠道弥漫型:息肉分布于全消化道,以反复上消化道出血为主要症状;多在儿童和青少年发病,恶变率较高[39,51-54]。
约有11% ~15%的FJPS患者可并发先天性疾病[51,54],如杵状指(趾)、肥大性肺性骨关节病、脑积水、唇裂、腭裂、先天性心脏病、肠旋转不良、脐疝、隐睾和美克尔憩室等。另外,FJPS患者也可伴发结直肠外肿瘤,如胃癌、十二指肠癌、胰腺癌等。
3.临床诊治现状:(1)诊断标准:目前临床多采用Jass诊断标准[53],结直肠幼年性息肉数目≥5枚;全胃肠道有幼年性息肉;不论幼年性息肉数目,有家族史者。(2)临床治疗:治疗关键是清除胃肠道息肉、防止并发症发生。手术结合内镜治疗是目前FJPS的主要临床治疗手段。对于小的、带蒂息肉应尽可能行内镜下灼除或圈套。对于反复便血、严重贫血、营养不良或息肉出现严重并发症无法用内镜摘除的患者,需考虑手术治疗。手术原则是切除全部病变肠管,但应尽可能保留肛门括约肌功能。
(三)PTEN错构瘤肿瘤综合征
PHTS是一组由PTEN基因突变而引起的常染色体显性遗传病[55]。其中具有结直肠息肉病表现的有:Cowden综合征(Cowden syndrome,CS又称多发性错构瘤综合征multiply hamartoma syndrome,MHS)和 Bannayan-Riley-Ruvalcaba综合征(BRRS综合征)。
1.Cowden综合征:(1)遗传学基础[56-58]:Cowden 综合征发病率约为1/200000,其遗传学基础是PETN基因突变;(2)临床病理特点:Cowden综合征是一种症状表现为结直肠多发性错构瘤息肉病、面部小丘疹、肢端角化病和口腔黏膜乳头状瘤的综合征[50,55-59]。息肉主要分布于降结肠,多呈半球形、群生状,可与其它类型息肉并存。食管、胃、小肠可伴发丘疹样息肉。面、颈部多发性扁平隆起性小丘疹。口腔黏膜、牙龈多见细小的圆石样丘疹或疣状小丘疹。70% ~80%的病例伴有甲状腺和乳腺病变[60-61]。
2.Bannayan-Riley-Ruvalcaba综合征
BRRS综合征是一种由PTEN突变引起的、罕见的常染色体显性遗传病[62],以结直肠息肉病、大头畸形、脂肪瘤病、血管瘤病和生殖器着色斑病为主要的临床特征。过去认为BRRS综合征与Cowden综合征不同,但现在越来越多的证据表明BRRS综合征与Cowden综合征有等位基因[62-64],因此,BRRS综合征和Cowden综合征可能是同一种疾病的不同表现。
(四)遗传性混合息肉病综合征
HMPS是一种罕见的常染色体显性遗传病[65],其特征是腺瘤性息肉和幼年性息肉混合存在。1997年才被首次报道,病例数很少。
1.遗传学基础:越来越多的学者将HMPS的遗传学基础确定为BMPR1A的胚系突变[50,65-68]。由于 BMPR1A的胚系突变也是FJPS的遗传学基础,因此,有学者认为HMPS应属于FJPS的变异亚型。
2.临床病理特点:息肉数目少(<15枚),全大肠分布.具有腺瘤性息肉和增生性息肉相重叠的混合性组织学特点。主要病理类型有管状腺瘤、绒毛状腺瘤、扁平息肉、增生性息肉和不典型幼稚息肉。患者患结直肠癌风险增加,但并不增加患肠外肿瘤的概率[65-68]。
四、预防性治疗
目前,遗传性结直肠癌的临床治疗仍以手术为主,辅以内镜治疗。但它们都是局部的、被动的治疗手段,无法达到预防疾病发生、发展的作用。而基因突变进而导致病变发生、发展、恶变、转移的一系例生物学过程需要很长的时间,并有多种细胞因子、酶等参与其中。如果能在其发生发展过程中的关键环节加以阻断或抑制,就可能起到抑制遗传性结直肠癌的发生、延缓其发展的作用。
(一)分子靶向治疗
针对遗传性结直肠癌的分子靶点而设计的肿瘤治疗方案具有治疗特异性强、效果显著和不损伤正常组织的优点[69]。代表有以下几类。
1.环氧合酶-2(COX-2)选择性抑制剂:研究显示[10,47,70-72]:COX-2 在上述所有遗传性结直肠癌中均有高表达。因此,应用COX-2抑制剂将可以抑制上述遗传性结直肠癌的发展。美国FDA于1999年批准了选择性COX-2抑制剂用于FAP的辅助治疗。选择性COX-2抑制剂于20世纪90年代开始上市,第一代有尼美舒利、美洛昔康、塞来昔布、罗非昔布等,第二代有伐地昔布、帕瑞昔布、依托昔布等。
将COX-2作为上述遗传性结直肠癌治疗的新靶点具有广阔的应用前景。原因:(1)COX-2在肿瘤细胞中高表达而在正常细胞中不表达,是良好的肿瘤治疗靶点.(2)筛选COX-2相关抗肿瘤药物相对容易。大量COX-2抑制剂作为消炎止痛一线药物已使用多年,筛选的药物不必再进行临床实验。(3)可根据COX-2延伸寻找和前列腺素合成相关的新的治疗靶点。
2.mTOR信号通路抑制剂:研究发现:mTOR信号通路参与了HNPCC、腺瘤性息肉病和错构瘤息肉病的发生、发展等过程[73-75]。因此,抑制mTOR信号通路将可以抑制上述疾病的发生和发展。雷帕霉素(也称西罗莫司)是mTOR信号通路的第一代抑制剂[76],具有抗淋巴细胞增殖、抗肿瘤和抗真菌的作用。新一代的mTOR信号通路抑制剂特癌适、依维莫司已经开始应用于临床。
大分子mTOR信号通路抑制剂存在人工合成困难、药物不稳定、用药途径不便、价格昂贵、免疫抑制等缺点。近年来,人们发现并人工合成了许多小分子mTOR信号通路抑制剂,它们具有稳定性好、可完全人工合成、服用方便、价格便宜、副作用较小的优点。有些小分子mTOR信号通路抑制剂同时具有mTOR信号通路相关分子靶点的抑制功能,特别是PI3K的双重或多重抑制剂将比单靶点抑制剂有更大的临床治疗优势。其中 NVP-BEZ235、PKI587、PKI179、GSK2126458、AZD8055、WYE-354等小分子mTOR信号通路抑制剂已进入临床实验阶段。
3.EGFR、HER-2、VEGF及其受体的抑制剂:目前针对EGFR、HER-2、VEGF及其受体的抑制剂已经开始应用于结直肠癌的临床治疗[69]。但关于他们在上述遗传性结直肠癌中的作用的研究很少[77-80]。能否将 EGFR、HER-2、VEGF及其受体的抑制剂应用于遗传性结直肠肿瘤患者的治疗中尚需进一步研究。
(二)中药治疗
目前,鉴于分子靶向药物尚未在临床上普及。而中医对消化道息肉的认识和治疗有其独到之处,同时中药具有服用方便、价廉、副作用小的优点。因此,中药治疗为消化道息肉患者提供了一种不错的选择。
1.消化道息肉的中医辩证:中医视息肉为痰凝瘀积之赘生物,将消化道息肉的临床表现归属于“肠癖”、“肠覃”、“泄泻”、“便血”等病证范畴。认为先天禀赋因素及饮食因素是消化道息肉形成的主要病因[81]。
2.消化道息肉的中药治疗:在中药治疗消化道息肉的资料中,以古方“济生乌梅丸”的使用频率最高。该方是宋代用于疗“肠风下血”而设。清朝人陈修园所著《时方歌括》中记载:下血淋漓治颇难,济生遗下乌梅丸,僵蚕炒研乌梅捣,醋下几回病即安(即僵蚕一两炒,乌梅肉一两半,共为末。醋糊丸桐子大,每服40~50丸,空心醋汤下)。此方为重庆中医研究所龚志贤医生治疗各种息肉的经验之方[82],其用于各种息肉患者的治疗疗效可靠。
济生乌梅丸的主药是乌梅和僵蚕[83]。制法为乌梅1500 g(选用肥大肉多的乌梅为上,酒醋浸泡一宿,去核、焙焦存性)、僵蚕500 g(米拌炒微黄为度)、穿山甲50 g(用碱水或皂水洗净,晒干,再用滑石粉入锅炒至甲片黄色鼓起为度,筛去滑石粉,放凉,碾粉)上药共为细末,炼蜜为丸,每丸重9 g,每日3次口服。制成后的丸药装入玻璃瓶内,放于干燥通风处,以防受潮霉烂变质。霉变不可服用。由于遗传性结直肠息肉好发于小儿,服丸药较困难,可改用乌梅、僵蚕各15 g煎汤,每日2次口服。
注意事项:服药期间饮食宜清淡,多食水果和蔬菜,保持大便通畅,忌食煎炒辛辣,忌烟酒。
3.中药灌肠:“五倍子乌梅汤”保留灌肠具有清湿热、祛腐化瘀、广谱抗炎之功效,可以延缓消化道息肉的发生发展[84]。方中主药五倍子、乌梅能收敛止血、平胬去腐;黄连、金银花、紫草、丹参有清热除湿解毒、活血化瘀之功;白芨有收敛止血、消肿生肌之功效,局部应用可促进创伤愈合;薄荷有清热作用;僵蚕有平胬去腐之功。
配伍及制法:五倍子10 g,乌梅15 g,黄连10 g,金银花10 g,紫草 15 g,白芨 15 g,薄荷 10 g,丹参 10 g,僵蚕 10 g,加水500 mL,水煎浓缩至100~150 mL,滤除杂质后装瓶备用。每晚睡前1 h点滴法保留灌肠1次,12天为一疗程。一疗程结束后休息7天,半年后重复第二疗程。
五、展望
目前临床对于遗传性结直肠癌尚无法进行病因治疗,如何达到提前预防、延缓息肉和癌肿生长、消灭息肉和癌肿是今后临床科研努力的主要方向和目标。研究发现:遗传性结直肠癌患者的消化道息肉和癌肿的形成需要很长的时间,多种细胞因子、酶等参与其中。如果能在其发生、发展途径中的关键环节加以阻断或抑制,那么就可以抑制遗传性结直肠癌的息肉和癌肿的形成、延缓其发展。分子靶向治疗将为遗传性结直肠癌患者开辟一个新的治疗途径。
[1] Kim JC,Kim SY,Roh SA,et al.Gene expression profiling:Canonical molecular changes and clinicopathological features in sporadic colorectal cancers.World JGastroenterol,2008,14(43):6662-6672.
[2] 陈明清,珠珠,戴莉萍,等.云南省遗传性大肠癌组织库的建立及管理.世界华人消化杂志,2008,16(27):3122-3125.
[3] 韩英.遗传性大肠癌的临床研究现状及进展.临床内科杂志,2007,24(8):519-520.
[4] 顾国利,周晓武,王石林.遗传性非息肉病性大肠癌的研究进展.世界华人消化杂志,2007,15(29):3115-3121.
[5] Castells A,Balaguer F,Castellví-Bel S,et al.Identification of Lynch syndrome:How should we proceed in the 21stcentury?World JGastroenterol,2007,13(33):4413-4416.
[6] Shen XS,Zhao B,Wang ZJ.Clinical features and hMSH2/hMLH1 germ-line mutations in Chinese patients with hereditary nonpolyposis colorectal cancer.Chin Med J,2008,121(14):1265-1268.
[7] Meyer LA,Broaddus RR,Lu KH.Endometrial cancer and Lynch syndrome:clinical and pathologic considerations.Cancer Control,2009,16(1):14-22.
[8] 顾国利,魏学明,王石林,等.遗传性非息肉病性大肠癌中hMSH2,hMLH1,TβRⅡ,MMP-7 及 TIMP-2 的表达和其特殊生物学行为间的关系.世界华人消化杂志,2007,15(15):1738-1744.
[9] 顾国利,魏学明,王石林,等.遗传性非息肉病性大肠癌组织E-cad,β-cat和MMP-7表达在侵袭转移中的作用.世界华人消化杂志,2007,15(18):2031-2036.
[10] 顾国利,王石林,魏学明,等.COX-2、β-cat、MMP-7 表达与遗传性非息肉病性大肠癌特殊侵袭转移行为的关系.世界华人消化杂志,2009,17(2):151-157.
[11] Jass JR.Hereditary non-polyposis colorectal cancer:The rise and fall of a confusing term.World J Gastroenterol,2006,12(31):4943-4950.
[12] Gylling A,Abdel-Rahman WM,Juhola M,et al.Is gastric cancer part of the tumour spectrum of hereditary non-polyposis colorectal cancer?A molecular genetic study.Gut,2007,56(7):926-933.
[13] 全国遗传性大肠癌协作组.中国人遗传性大肠癌筛检标准的实施方案.中华肿瘤杂志,2004,26(3):191-192.
[14] Herráiz M,Muňoz-Navas M.Recognition and management of hereditary colorectal cancer syndromes.Rev Esp Enferm Dig,2009,101(2):125-132.
[15] Koornstra JJ,Mourits MJ,Sijmons RH,et al.Management of extracolonic tumours in patients with Lynch syndrome.Lancet Oncol,2009,10(4):400-408.
[16] Kanter-Smoler G,Fritzell K,Rohlin A,et al.Clinical characterization and the mutation spectrum in Swedish adenomatous polyposis families.BMCMed,2008,6:10.
[17] 冯莉芳,赖仁胜,刘丽,等.APC基因MCR区突变与大肠肿瘤患者临床发病的关系.世界华人消化杂志,2009,17(5):532-537.
[18] Wachsmannova-Matelova L,Stevurkova V,Adamcikova Z,et al.Different phenotype manifestation of familial adenomatous polyposis in families with APC mutation at codon 1309.Neoplasma,2009,56(6):486-489.
[19] Cai SR,Zhang SZ,Zheng S.Clinical features of familial adenomas polyps in Chinese and establishment of its immortal lymphocyte cell lines.World J Gastroenterol,2007,13(20):2858-2861.
[20] 苏芳,王涛,王邦茂.衰减型家族性腺瘤性息肉病.中国消化内镜杂志,2008,2(8):20-26.
[21] Sieber OM,Segditsas S,Knudsen AL,et al.Disease severity and genetic pathways in attenuated familial adenomatous polyposis vary greatly but depend on the site of the germline mutation.Gut,2006,55(10):1440-1448.
[22] 杨邵瑜,蔡善荣,张苏展.家族性腺瘤性息肉病及其亚型的临床及遗传表型.实用肿瘤杂志,2007,22(3):270-273.
[23] Nielsen M,Hes FJ,Nagengast FM,et al.Germline mutations in APC and MUTYH are responsible for the majority of families with attenuated familial adenomatous polyposis.Clin Genet,2007,71(5):427-433.
[24] Gómez-Fernández N,Castellví-Bel S,Fernández-Rozadilla C,et al.Molecular analysis of the APC and MUTYH genes in Galician and Catalonian FAP families:a different spectrum of mutations?BMC Med Genet,2009,10:57.
[25] Poulsen ML,Bisgaard ML.MUTYH Associated Polyposis(MAP).Curr Genomics,2008,9(6):420-435.
[26] 周卉卉,吕炳建,来茂德.遗传性MUTYH基因突变与结直肠癌.浙江大学学报(医学版),2007,34(4):406-411.
[27] Nielsen M,Joerink-van de MC,Jones N,et al.Analysis of MUTYH genotypes and colorectal phenotypes in patients With MUTYH-associated polyposis.Gastroenterology,2009,136(2):471-476.
[28] Smud D,Augustin G,Kekez T,et al.Gardner's syndrome:Genetic testing and colonoscopy are indicated in adolescents and young adults with cranial osteomas :A case report.World J Gastroenterol,2007,13(28):3900-3903.
[29] Lipton L,Tomlinson I.The genetics of FAP and FAP-like syndromes.Fam Cancer,2006,5(3):221-226.
[30] Fotiadis C,Tsekouras DK,Antonakis P,et al.Gardner’s syndrome:A case report and review of the literature.World J Gastroenterol,2005,11(34):5408-5411.
[31] Gu GL,Wang SL,Wei XM,et al.Diagnosis and treatment of Gardner syndrome with gastric polyposis:a case report and review of the literature.World J Gastroenterol,2008,14(13):2121-2123.
[32] Reuss D,von Deimling A.Hereditary tumor syndromes and gliomas.Recent Results Cancer Res,2009,171:83-102.
[33] Lebrun C,Olschwang S,Jeannin S,et al.Turcot syndrome confirmed with molecular analysis.Eur J Neurol,2007,14(4):470-472.
[34] Sjursen W,Bjφrnevoll I,Engebretsen LF,et al.A homozygote splice site PMS2 mutation as cause of Turcot syndrome gives rise to two different abnormal transcripts.Fam Cancer,2009,8(3):179-186.
[35] Sarin S,Bernath A.Turcot syndrome(glioma polyposis):a case report.South Med J,2008,101(12):1273-1274.
[36] Wuthrich P,Gervaz P,Ambrosetti P,et al.Functional outcome and quality of life after restorative proctocolectomy and ileo-anal pouch anastomosis.Swiss Med Wkly,2009,139(13-14):193-197.
[37] Edlich R,Cross CL,Wack CA,et al.Revolutionary advances in the diagnosis and treatment of Familial Adenomatous Polyposis.J Environ Pathol Toxicol Oncol,2009,28(1):47-52.
[38] Filho OG,Gordan AN,Mello Rde A,et al.Myoid hamartomas of the breast:report of 3 cases and review of the literature.Int JSurg Pathol,2004,12(2):151-153.
[39] Calva D,Howe JR.Hamartomatous polyposis syndromes.Surg Clin North Am,2008,88(4):779-817.
[40] 王石林,顾国利.Peutz-Jeghers综合征临床诊断治疗的现状和相关问题.世界华人消化杂志,2008,16(21):2385-2389.
[41] Li LJ,Wang ZQ,Wu BP.Peutz-Jeghers syndrome with small intestinal malignancy and cervical carcinoma.World J Gastroenterol,2008,14(48):7397-7399.
[42] Hezel AF,Bardeesy N.LKB1,linking cell structure and tumor suppression.Oncogene,2008,27(55):6908-6919.
[43] Fan D,Ma C,Zhang H.The molecular mechanisms that underlie the tumor suppressor function of LKB1.Acta Biochim Biophys Sin(Shanghai),2009,41(2):97-107.
[44] Vasovcák P,Puchmajerová A,Roubalík J,et al.Mutations in STK11 gene in Czech Peutz-Jeghers patients.BMC Med Genet,2009,10:69.
[45] Amos CI,Keitheri-Cheteri MB,Sabripour M,et al.Genotypephenotype correlations in Peutz-Jeghers syndrome.J Med Genet,2004,41(5):327-333.
[46] 王石林,毛高平,顾国利.Peutz-Jeghers综合征胃肠道息肉的36例诊治经验.中华胃肠外科杂志,2009,12(4):428.
[47] 王石林,顾国利.Peutz-Jeghers综合征胃肠道息肉的药物干预性治疗的进展.中国普外基础与临床杂志,2009,16(4):333-335.
[48] 宁守斌,毛高平,曹传平,等.双气囊小肠镜对Peutz-Jeghers综合征患者小肠息肉的治疗价值.世界华人消化杂志,2008,16(14):1588-1591.
[49] 毛高平,宁守斌,白莉,等.双气囊电子小肠镜在小肠疾病诊断中的应用价值.世界华人消化杂志,2007,15(28):3049-3053.
[50] Chen HM,Fang JY.Genetics of the hamartomatous polyposis syndromes:a molecular review.Int JColorectal Dis,2009,24(8):865-874.
[51] Chow E,Macrae F.A review of juvenile polyposis syndrome.J Gastroenterol Hepatol,2005,20(11):1634-1640.
[52] 姚蓝,宋家武.肠息肉发生的细胞和分子生物学研究进展.世界华人消化杂志,2006,14(30):2958-2961.
[53] 刘剑,李群,陈丽荣,等.家族性幼年性息肉病的家系研究.中华普通外科杂志,2006,21(8):553-556.
[54] 王丽君,宁建文,季峰,等.成人胃幼年性息肉和幼年性息肉病临床病理特征及癌变趋势探讨.中华消化杂志,2005,25(1):43-44.
[55] Blumenthal GM,Dennis PA.PTEN hamartoma tumor syndromes.Eur J Hum Genet,2008,16(11):1289-1300.
[56] Umemura K,Takagi S,Ishigaki Y,et al.Gastrointestinal polyposis with esophageal polyposis is useful for early diagnosis of Cowden's disease.World J Gastroenterol,2008,14(37):5755-5759.
[57] 李季,田素礼,李巍,等.结直肠癌中PTEN的缺失表达及临床意义.世界华人消化杂志,2006,14(28):2771-2775.
[58] 刘玉环,王锡智,蔡在龙,等.PTEN基因突变与遗传性癌症综合征及其相关疾病的研究进展.第二军医大学学报,2005,26(1):90-92.
[59] Blanco V,Keochgerián V.Cowden's syndrome:Case report,with reference to an affected family.Med Oral Patol Oral Cir Bucal,2006,11(1):E12-E16.
[60] Pilarski R.Cowden syndrome:a critical review of the clinical literature.JGenet Couns,2009,18(1):13-27.
[61] LeoãJC,Batista V,Guimarães PB,et al.Cowden's syndrome affecting the mouth,gastrointestinal,and central nervous system:a case report and review of the literature.Oral Surg Oral Med Oral Pathol Oral Radiol Endod,2005,99(5):569-572.
[62] Orloff MS,Eng C.Genetic and phenotypic heterogeneity in the PTEN hamartoma tumour syndrome.Oncogene,2008,27(41):5387-5397.
[63] Lachlan KL,Lucassen AM,Bunyan D,et al.Cowden syndrome and Bannayan Riley Ruvalcaba syndrome represent one condition with variable expression and age-related penetrance:results of a clinical study of PTEN mutation carriers.J Med Genet,2007,44(9):579-585.
[64] Pezzolesi MG,Platzer P,Waite KA,et al.Differential expression of PTEN-targeting microRNAs miR-19a and miR-21 in Cowden syndrome.Am JHum Genet,2008,82(5):1141-1149.
[65] 彭慧,曹霞,余光荣,等.华人遗传性混合息肉病综合征BMPR1A基因种系突变检测.中山大学学报(医学科学版),2005,26(3):316-320.
[66] Cao X,Eu KW,Kumarasinghe MP,et al.Mapping of hereditary mixed polyposis syndrome(HMPS)to chromosome 10q23 by genomewide high-density single nucleotide polymorphism(SNP)scan and identification of BMPR1A loss of function.JMed Genet,2006,43(3):e13.
[67] Sweet K,Willis J,Zhou XP,et al.Molecular classification of patients with unexplained hamartomatous and hyperplastic polyposis.JAMA,2005,294(19):2465-2473.
[68] 彭慧,曹霞,李慧华,等.华人遗传性混合息肉病综合征患者单倍型及遗传连锁分析.中华胃肠外科杂志,2005,8(4):316-319,324.
[69] 魏学明,顾国利,任力,等.大肠癌 EGFR、HER-2、VEGF表达特点及其对分子靶向治疗的指导意义.世界华人消化杂志,2009,17(18):1836-1841.
[70] Takeda H,Miyoshi H,Tamai Y,et al.Simultaneous expression of COX-2 and mPGES-1 in mouse gastrointestinal hamartomas.Br J Cancer,2004,90(3):701-704.
[71] Brazowski E,Misonzhnick-Bedny F,Rozen P.Cyclooxygenase-2 expression in the hereditary mixed polyposis syndrome.Dig Dis Sci,2004,49(11-12):1906-1911.
[72] van Hattem WA,Brosens LA,Marks SY,et al.Increased cyclooxygenase-2 expression in juvenile polyposis syndrome.Clin Gastroenterol Hepatol,2009,7(1):93-97.
[73] Rosner M,Hanneder M,Siegel N,et al.The mTOR pathway and its role in human genetic diseases.Mutat Res,2008,659(3):284-292.
[74] Wei C,Amos CI,Zhang N,et al.Suppression of Peutz-Jeghers polyposis by targeting mammalian target of rapamycin signaling.Clin Cancer Res,2008,14(4):1167-1171.
[75] Fujishita T,Aoki K,Lane HA,et al.Inhibition of the mTORC1 pathway suppresses intestinal polyp formation and reduces mortality in ApcDelta716 mice.Proc Natl Acad Sci USA,2008,105(36):13544-13549.
[76] Höpfner M,Schuppan D,Scherübl H.Growth factor receptors and related signalling pathways as targets for novel treatment strategies of hepatocellular cancer.World J Gastroenterol,2008,14(1):1-14.
[77] Roberts RB,Min L,Washington MK,et al.Importance of epidermal growth factor receptor signaling in establishment of adenomas and maintenance of carcinomas during intestinal tumorigenesis.Proc Natl Acad Sci USA,2002,99(3):1521-1526.
[78] Brugarolas J,Kaelin WG.Dysregulation of HIF and VEGF is a unifying feature of the familial hamartoma syndromes.Cancer Cell,2004,6(1):7-10.
[79] McGarrity TJ,Peiffer LP,Billingsley ML.Overexpression of epidermal growth factor receptor in Peutz-Jeghers syndrome.Dig Dis Sci,1999,44(6):1136-1141.
[80] Ylikorkala A,Rossi DJ,Korsisaari N,et al.Vascular abnormalities and deregulation of VEGF in Lkb1-deficient mice.Science,2001,293(5533):1323-1326.
[81] 高家信.大肠息肉的中医临床研究概况.现代中西医结合杂志,2001,10(2):117-118.
[82] 李志,陈原,张国英.龚志贤加味济生乌梅丸临床应用举隅.中国中医急症杂志,2002,11(2):151.
[83] 杨炜,徐进康.乌梅用于大肠息肉治疗探讨.中国民族民间医药杂志,2010,19(4):45-48.
[84] 张凤清,方军.肠息肉摘除术后中药灌肠预防复发的疗效观察及护理.护士进修杂志,2002,17(3):200-201.
丁振,顾国利.遗传性结直肠癌的体系结构和诊治进展[J/CD].中华结直肠疾病电子,2013,2(4):184-190.
猜你喜欢
中国现代医生(2022年19期)2022-11-04
中国肿瘤临床(2022年14期)2022-08-09
中国临床医学影像杂志(2022年2期)2022-05-25
昆明医科大学学报(2022年4期)2022-05-23
中国听力语言康复科学杂志(2021年6期)2021-12-21
天津医科大学学报(2021年2期)2021-03-29
中国临床医学影像杂志(2019年5期)2019-08-27
中国临床医学(2019年2期)2019-05-21
中国内镜杂志(2017年2期)2017-03-20
中西医结合心血管病杂志(电子版)(2017年1期)2017-01-11
