
MYCT1新转录本的克隆与表达分析
2014-03-21 07:42符爽孙开来富伟能
中国医科大学学报 2014年5期
符爽,孙开来,富伟能
(中国医科大学1.基础医学院遗传学教研室,沈阳110001;2.附属盛京医院血液研究室,沈阳110022)
MYCT1新转录本的克隆与表达分析
符爽1,2,孙开来1,富伟能1
(中国医科大学1.基础医学院遗传学教研室,沈阳110001;2.附属盛京医院血液研究室,沈阳110022)
目的确定MYCT1基因的转录起始点并克隆该基因新的转录本,探讨该基因的结构和功能。方法利用已知MYCT1的保守序列,应用5′-RACE技术确定转录起始点位置,结合3′-RACE拼接得到新转录本的精确全长;然后利用生物信息学软件对新转录本与MYCT1的cDNA序列及氨基酸序列进行对比分析;最后利用RT-PCR的方法分析新转录本的细胞表达谱。结果成功克隆得到长1 106 bp的新转录本MYCT1-TV,其转录起始点位于ATG上游140 bp处。MYCT1-TV与MYCT1在结构上无明显差异,并广泛表达于各个细胞系。结论MYCT1转录起始点的确定和新转录本MYCT1-TV的克隆,为进一步研究MYCT1基因的转录调控机制及基因功能奠定了实验基础。
MYCT1;MYCT1-TV;喉癌;RACE
MYCT1(Myc target 1)是本研究室前期应用实体瘤培养、常规G显带、FISH及电子杂交的方法从喉鳞状细胞癌(laryngeal squamous cell carcinoma,LSCC,以下简称喉癌)中克隆得到的新基因,曾命名MTLC(c-Myc target from laryngeal cancer cells),Gen-Bank登录号为AF_527367[1]。
MYCT1基因定位于染色体6q25,全长约21 kb,含2个外显子,其mRNA序列长约1 006 bp,编码含235个氨基酸的蛋白质。本研究组在前期研究中通过生物信息学方法,预测该基因转录起始点位于ATG上游47 bp处[1],但未经实验证实。进一步研究MYCT1基因的结构并确定其转录起始点,对了解该基因的功能和阐明其转录调控机制具有重要意义。本研究利用已知MYCT1的保守序列,以正常人外周血RNA为模板,应用5′-RACE(5′-rapid amplification of cDNA ends)的方法确定了MYCT1的转录起始点,并结合3′-RACE克隆得到MYCT1新的转录本,通过生物信息学的方法对比分析2个转录本的结构,并检测新转录本在不同细胞系中的表达情况,为明确MYCT1的结构和功能奠定了实验基础。
1 材料与方法
1.1 实验材料与试剂
细胞系:人喉癌Hep2细胞、人胚肾HEK293细胞、人肝癌Bel7402细胞、人宫颈癌HeLa细胞、人高分化胃癌BGC823细胞、人中分化胃癌SGC7901细胞、人低分化胃癌MKN1细胞、人胃黏膜上皮GES1细胞(购自中科院上海细胞库)。
主要试剂:反转录试剂盒、胶回收试剂盒、pMD18-T载体、JM109感受态细胞及PCR相关试剂购自TaKaRa公司,全血RNA提取试剂盒购自Galen Biopharm公司,无内毒素质粒小提中量试剂盒购自Tiangen公司,SMARTTMRACE cDNA Amplification Kit购自Clontech公司。
培养基:LB培养基(自配),其中胰蛋白胨和酵母提取物为Sigma公司产品,RPMI1640培养基、胎牛血清为Gibco公司产品。
1.2 实验方法
1.2.1 全血RNA的提取及RNA质量的鉴定:取2 mL正常人新鲜抗凝血加入6 mL红细胞裂解液1× RLB,颠倒混匀。室温放置10 min,期间可轻弹颠倒混匀。离心后弃红色上清,留下完整白细胞团在管底。按Galen Biopharm公司试剂盒说明书步骤进行全血RNA的提取。将电泳槽冲洗干燥后用0.1% DEPC处理水浸泡过夜,取2 μL加样缓冲液与3 μL RNA溶液混匀后于l%的琼脂糖凝胶电泳,4℃冰箱中130 V电泳27 min。
1.2.2MYCT1基因转录起始点的确定及新转录本的克隆:以MYCT1的保守序列为模板,按照RACE试剂盒说明书要求设计模板特异性引物(GSP)和巢式PCR引物(NGSP)。引物序列如下:GSP1:5′-CATAGGCAGGTGGGGGCGGTGTT-3′;GSP2:5′-TATCCATGGCAATCGGGCTGGTACT-3′;NGSP1:5′-TGGACCAGTAGTCAGGACGGCTCAGA-3′;NGSP2:5′-AGTGGCTGTGAACGTCGAAGCAACC-3′。然后分别进行5′-和3′-RACE扩增,分别以设计的GSP1、GSP2为5′-RACE和3′-RACE的特异引物,与UPM(试剂盒自带)进行cDNA 5′端和3′端的一次扩增,然后分别以NGSP1、NGSP2为巢式PCR的特异引物,与NUP(试剂盒自带)进行cDNA 5′端和3′端的二次扩增。分别将5′端和3′端的扩增产物回收纯化后,克隆至pMD18-T载体,转化大肠杆菌JM109感受态细胞,挑选阳性克隆送南京金斯瑞生物技术有限公司测序。
1.2.3 RT-PCR验证RACE结果:为了排除RACE受基因组污染的可能性,采用RT-PCR的方法对RACE结果的真实性进行验证。设计上下游引物扩增拼接后得到的cDNA全长,以5′-RACE cDNA为模板进行PCR扩增。引物序列如下:Forward,5′-GAACAC AAGTATATGAGGGGTTG-3′;Reverse,5′-AGTCAC ATTGGAAGTAAATGAGGATTG-3′,反应体系与说明书一致。
PCR反应条件为:95℃4 min→(95℃30 s、60℃15 s、72℃2 min20 s)×35 cycles→72℃7 min→4℃∞。将扩增产物回收纯化,经连接转化后,挑取阳性克隆送南京金斯瑞生物技术有限公司测序。
1.2.4 RT-PCR实验检测MYCT1新转录本在各细胞系中的表达水平:分别收集5×106个Bel7402、Hep2、HeLa、BGC823、SGC7901、MKN1、GES1和HEK293细胞,用Trizol方法提取细胞总RNA,按照试剂盒说明书反转录成cDNA。以上述提取的各细胞系的总cDNA为模板,以正常人外周血中MYCT1-TV的mRNA表达水平作为阳性对照,通过PCR的方法检测MYCT1-TV在各细胞系的表达水平。MYCT1-TV引物序列:Forward,5′-TAATAACACAACAAGTTTA GGGAGT-3′;Reverse,5′-AGTCACATTGGAAGTAA ATGAGGATTG-3′,产物大小为929 bp。β-actin引物序列:Forward,5′-CTCTTCCAGCCTTCCTTCCT-3′;Reverse,5′-CACCTTCACCGTTCCAGT TT-3′,产物大小为511 bp。
2 结果
2.1MYCT1新转录本的获得及结构分析
2.1.1 总RNA的鉴定结果:从外周血提取的总RNA经1%琼脂糖凝胶电泳可见28 S、18 S和5.8 S rRNA 3条清晰的带(图1),OD260/OD280的比值约为1.90,浓度约为0.2 g/L,表明抽提的RNA质量较好,可以用于后续实验。
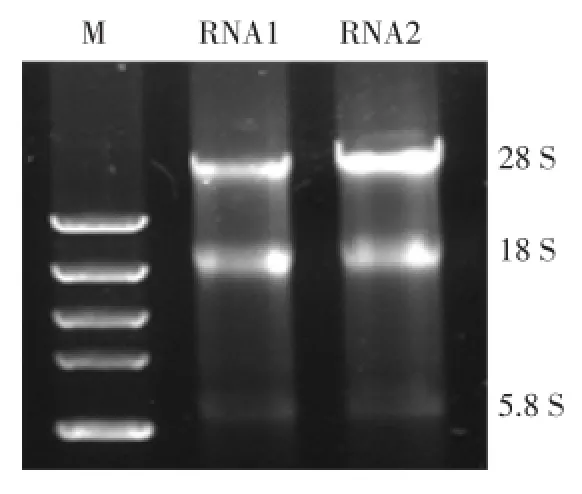
图1 RNA完整性电泳图Fig.1 Agar electrophoresis of RNA integrity
2.1.2 5′/3′-RACE巢式PCR检测结果:5′端经巢式PCR扩增得到长约690 bp和1 400 bp的2个DNA片段,3′端经巢式PCR扩增得到长约750 bp、1 200 bp、1 800 bp、2 500 bp和3 000 bp的5个DNA片段(图2)。
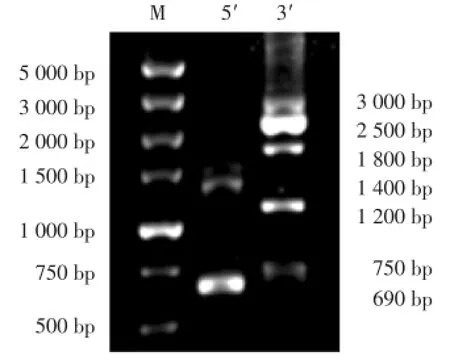
图2 巢式PCR产物电泳检测结果Fig.2 Result from electrophoresis detection of nested PCR products
2.1.3MYCT1全长cDNA的拼接及转录起始点的鉴定结果:将巢式得到的7个片段经过连接转化后送测序鉴定。根据测序结果,我们将5′序列和3′序列进行了拼接,并与Blast进行比对,结果得到1个全长为1 106 bp的MYCT1新的转录本(图3),命名为MYCT1-TV(myc target 1 transcript variant 1),并提交GenBank数据库,获得GenBank登录号GU997693,并确定此转录本的转录起始点为cDNA的第一个碱基“G”。

图3 拼接后的MYCT1⁃TV cDNA序列Fig.3 MYCT1⁃TV cDNA sequence after splicing
2.1.4 拼接后MYCT1-TVcDNA序列鉴定结果:参照我们所设计的PCR引物,RT-PCR产物大小理论上应为1 074 bp(除去polyA尾长度)。经RT-PCR检测,我们获得大小为1 000 bp左右的单一条带(图4),与预期相符,将产物胶回收后克隆至pMD18-T载体,连接、转化后,筛选阳性质粒送测序,提取插入序列进行Blast分析,显示克隆的片段确系MYCT1-TV的cDNA序列。
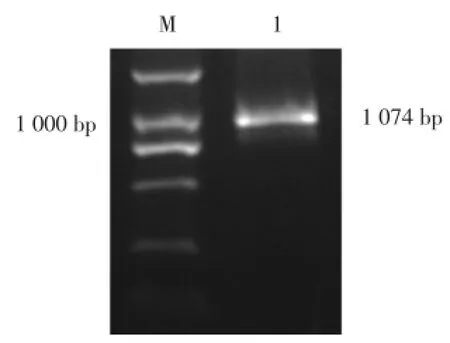
图4 RT⁃PCR拼接后MYCT1⁃TV cDNA电泳图Fig.4 Agar electrophoresis of MYCT1⁃TV cDNA after splicing by RT⁃PCR
2.1.5MYCT1-TV与MYCT1序列的比较分析:应用ExPASy proteomics server软件,我们对MYCT1-TV的cDNA序列进行了分析,结果如图5所示:MYCT1-TVcDNA全长1 106 bp,编码包含187个氨基酸的蛋白质,蛋白分子量为20 835 Da,等电点为10.26,5′UTR长140 bp,3′UTR长370 bp,polyA尾长32 bp,其转录起始点位于MYCT1-TV的翻译起始点ATG上游140 bp处(图6A)。Blast分析结果显示:MYCT1-TV-cDNA 5′旁侧序列比MYCT1短,3′旁侧序列比MYCT1长(图6A);蛋白质序列的比较分析发现,MYCT1-TV编码的氨基酸序列除了比MYCT1少48个氨基酸序列外,其余187个氨基酸序列与后者完全一致(图6B),且经ExPASy proteomics server的分析结果显示,减少的这48个氨基酸无明显的结构域和功能域。

图5 MYCT1⁃TV cDNA序列特征Fig.5 cDNA characteristics of MYCT1⁃TV
2.2MYCT1-TV在各细胞系中表达水平的检测结果
RT-PCR结果显示:MYCT1-TVmRNA在各细胞系中均有表达,在GES1和HEK293等正常细胞系中的表达水平显著高于Hep2、HeLa、BGC823、SGC7901、Bel7402和MKN1等癌细胞系(P<0.01,图7),MYCT1-TVmRNA在GES1和HEK293细胞系的表达水平与正常人外周血细胞相比无显著差异(P>0.05,图7),且在各肿瘤细胞中的表达也无显著差异(P>0.05,图7)。

图6 MYCT1⁃TV和MYCT1的序列比较Fig.6 Comparison of MYCT1⁃TV and MYCT1sequences

图7 MYCT1⁃TV在人各细胞系中mRNA的表达水平Fig.7 MYCT1⁃TV mRNA levels in different human cell lines
3 讨论
RACE是基于PCR技术,由已知的部分cDNA序列来获得完整cDNA序列的一种方法,为Frohman在1985年首次报道[2]。该技术的出现解决了PCR扩增效率低和特异性差,不易得到完整的全长基因的问题。它可以从低丰度的转录本中快速扩增cDNA的 5′和3′末端。近年来对RACE技术进行了改进[3~5],SMART-RACE技术就是其中最经典、应用最广泛的技术之一[6~10]。SMART技术最大限度地提高了在反转录反应中获取全长cDNA的可能,只需一种引物和基因特异引物配对即可扩增出5′和3′RACE产物,而且只需cDNA第一链为模板,无需进行cDNA第二链的合成,也无需接头的连接。对于只有少量起始材料的RNA,SMART-RACE也可以通过应用长距离PCR的方法,利用含poly(A)的RNA或总RNA甚至低丰度的转录产物作为起始材料来克隆全长cDNA[11],已成为目前公认的确定基因转录起始点、克隆基因cDNA精确全长的经典方法。
本研究利用GenBank数据库中MYCT1的保守序列,通过SMART-RACE方法,从正常人外周血RNA中克隆获得长1 106 bp的MYCT1的新的转录本MYCT1-TV,并精确定位了该基因的转录起始点位于MYCT1-TV基因ATG上游140 bp处。MYCT1-TV具有完整的开放阅读框,且3′端有poly(A)加尾。开放阅读框分析表明,该转录本编码含187个氨基酸的蛋白质。氨基酸序列的比较结果发现,MYCT1-TV编码的187个氨基酸完全是MYCT1编码的235个氨基酸的一部分,且少的这48个氨基酸无明显的结构域和功能域。表明2个转录本在结构上无明显差异,但它们在功能上是否存在差异有待实验进一步证实。
基因表达谱分析表明,MYCT1-TV在各个细胞系中均有表达,表明MYCT1-TV基因并不是喉部组织特异性基因,在其他组织细胞中也有表达,该基因可能对维持细胞的正常生理功能是必需的。另外,MYCT1-TV在肿瘤细胞中的表达水平显著低于其在正常细胞系中的表达水平,这与MYCT1在喉癌[12]、胃癌[13]和乳腺癌[14]等肿瘤组织中表达下调的结果一致,表明其可能同样发挥肿瘤抑制基因功能。
总之,MYCT1-TV基因转录起始点的确定和cDNA全长的克隆为研究该基因启动子区的调控和基因功能奠定了基础。
[1]邱广斌,邱广蓉,徐振明,等.6q25区域内一个新基因MTLC的克隆及特性分析[J].中华医学遗传学杂志,2003,20(2):94-97.
[2]徐烨,刘雅婷,代文琼,等.几种主要的RACE技术及应用[J].中国农业科技导报,2012,14(2):81-87.
[3]Yeku O,Frohman MA.Rapid amplification of cDNA ends(RACE)[J].Methods Mol Biol,2011,703:107-122.
[4]Dallmeier K,Neyts J.Simple and inexpensive three-step rapid amplification of cDNA 5′ends using 5′phosphorylated primers[J]. Anal Biochem,2013,434(1):1-3.
[5]Bower NI,Johnston IA.Targeted rapid amplification of cDNA ends(T-RACE)-an improved RACE reaction through degradation of nontarget sequences[J].Nucleic Acids Res,2010,38(21):e194.
[6]Li G,Luo H,Sun G,et al.Cloning and characterization of a novel apolipoprotein gene,apolipoprotein AV,in tree shrews[J].Mol Biol Rep,2013,40(9):5429-5438.
[7]Rampias TN,Fragoulis EG,Sideris DC.Efficient cloning of alternatively polyadenylated transcripts via hybridization capture PCR[J]. Curr Issues Mol Biol,2012,14(1):1-8.
[8]Zeng LG,Wang JH,Li YJ,et al.Molecular characteristics and expression of calmodulin cDNA from the freshwater pearl mussel,Hyriopsis schlegelii[J].Genet Mol Res,2012,11(1):42-52.
[9]Sun G,Li M,Li H,et al.Molecular cloning and SNP association analysis of chicken PMCH gene[J].Mol Biol Rep,2013,40(8):5049-5055.
[10]Cheng H,Kong W,Hou D,et al.Isolation,characterization,and expression analysis of CmMLO2 in muskmelon[J].Mol Biol Rep,2013,40(3):2609-2615.
[11]Tabansky I,Nurminsky DI.Mapping of transcription start sites by direct sequencing of SMART RACE products[J].Biotechniques,2009,34(3):482,485-486.
[12]Qiu GB,Hao DM,Gong LG,et al.MTLC gene was down-regulated in laryngeal cancer tissues and could promote apoptosis of Hep2 cells[J].Chin J Lab Med,2005,28(11):1178-1180.
[13]Qiu GB,Gong LG,Hao DM,et al.Expression of MTLC gene in gastric carcinoma[J].World J Gastroenterol,2009,9(10):2160-2163.
[14]欧阳小明,黄世章,梅开勇.MTLC基因在乳腺癌组织中的表达及其意义[J].中国煤炭工业医学杂志,2006,9(7):695-696.
(编辑 王又冬)
Cloning and Expression Analysisofa Novel MYCT1Transcript
FUShuang1,2,SUNKai-lai1,FUWei-neng1
(1.DepartmentofMedicalGenetics,College ofBasic MedicalScience,China MedicalUniversity,Shenyang 110001,China;2.DepartmentofHematology Laboratory,Shengjing Hospital,China MedicalUniversity,Shenyang 110022,China)
ObjectiveTo identify the transcriptional start site and clone a novel transcript variant ofMYCT1(Myc target 1)for further study of its structure and function.MethodsTranscriptional start site was confirmed usingMYCT1conserved sequence by 5′-RACE method and a novelMYCT1isoform was cloned by splicing with 3′-RACE PCR product.Then,the cDNA or amino acid sequence betweenMYCT1and its isoform was compared using bioinformatics server.Finally,the expression profile of this novel transcript in different cell lines was detected through RT-PCR.Re⁃sultsA 1 106 bp transcript named MYCT1-TV(Myc target 1 transcript variant 1)was successfully cloned,and its transcriptional start site was confirmed which located at 140 bp upstream of the ATG start codon of MYCT1-TV.MYCT1-TV shows no obviously structural difference withMYCT1and is widely expressed in various cell lines.ConclusionThe transcriptional start site analysis and MYCT1-TV cloning provide an experimentalbasisforthe furtherexploration and understanding ofthe function and the transcriptionalregulation mechanism ofMYCT1.
MYCT1;MYCT1-TV;laryngeal squamous cell carcinoma;RACE
R394.3
A
0258-4646(2014)05-0388-05
国家自然科学基金(81372876);国家自然科学基金青年基金(81300420)
符爽(1983-),女,助理研究员,博士.
富伟能,E-mail:wnfu@mail.cmu.edu.cn
2014-01-06
网络出版时间:
猜你喜欢
环球时报(2022-09-20)2022-09-20
今日农业(2020年24期)2020-12-15
中国洗涤用品工业(2019年4期)2019-05-11
中成药(2017年3期)2017-05-17
小资CHIC!ELEGANCE(2015年14期)2015-09-23
小资CHIC!ELEGANCE(2015年15期)2015-09-01
医学研究杂志(2015年9期)2015-07-01
医学研究杂志(2015年9期)2015-07-01
癌变·畸变·突变(2015年4期)2015-02-27
中华胰腺病杂志(2014年1期)2014-08-04
