
弥漫性轴索损伤后内质网钙释放对轴突内早期钙离子浓度的影响
2014-07-18 11:53宋锦宁安吉洋李丹东马旭东赵雅慧
西安交通大学学报(医学版) 2014年6期
李 宇,宋锦宁,张 明,安吉洋,孙 鹏,李丹东,马旭东,赵雅慧
(西安交通大学医学院第一附属医院神经外科,陕西西安 710061)
◇专题研究◇
弥漫性轴索损伤后内质网钙释放对轴突内早期钙离子浓度的影响
李 宇,宋锦宁,张 明,安吉洋,孙 鹏,李丹东,马旭东,赵雅慧
(西安交通大学医学院第一附属医院神经外科,陕西西安 710061)
目的 研究弥漫性轴索损伤(diffuse axonal injury, DAI)后内质网(endoplasmic reticulum, ER)钙释放的变化,以及对轴突内早期钙离子浓度的影响,并探讨造成ER钙释放的可能分子机制。方法 使用体外培养7~12 d的小鼠神经元行轴突牵拉损伤建立体外DAI模型,根据是否行牵拉损伤分为损伤组及对照组。使用ER Tracker Red标记ER,观察ER在轴突损伤前后的分布。通过Fluo-4 AM钙探针及激光共聚焦钙成像技术测量单个轴突内钙离子浓度的变化,并使用20 mmol/L咖啡因作用于细胞,使ER内钙离子快速排空,测量轴突部位咖啡因作用前后的钙离子浓度变化,从而间接观察ER内钙存量的变化。结果 ER Tracker Red荧光结果显示ER存在于神经元轴突部位中,损伤后的轴突肿胀部位存在ER聚集的现象。神经元牵拉损伤后,被牵拉的轴突钙离子浓度明显升高,与对照组比较,差异有统计学意义(P<0.05),受损的轴突表现出肿胀及“串珠样”变性等病理学改变。除损伤后2 min外,使用无钙细胞外液虽然明显降低了牵拉损伤导致的轴突的钙离子浓度升高,但在2~30 min时间段中钙离子浓度升高依然存在,与对照组比较,差异有统计学意义(P<0.05)。对照组中未经牵拉损伤的神经元轴突可被20 mmol/L咖啡因激起一个明显的轴突钙离子浓度升高,而牵拉损伤后的轴突对咖啡因的这种钙升高反应程度明显下降,与对照组比较,差异有统计学意义(P<0.05)。结论 DAI后的轴突钙离子浓度升高不仅由细胞外钙内流引起,细胞内钙释放同样参与其中。ER是引起这种DAI后轴突内钙释放的细胞器来源。
脑外伤;弥漫性轴索损伤;钙;内质网
弥漫性轴索损伤(diffuse axonal injury, DAI)是脑外伤(traumatic brain injury, TBI)的一种重要的病理形式,也被认为是一种特殊的TBI损伤类型[1-2]。DAI不同于其他类型的TBI,是因为其生物力学机制并非直接的机械性暴力作用于头颅,而是机械暴力作用于头颅后导致的头颅加速性运动所致的剪应力损伤,最终剪应力作用于轴突(尤其是长轴突),从而导致广泛的轴突变性、断裂[3]。
目前的研究认为,DAI后的轴突变性及断裂过程主要发生在继发性损伤阶段(secondary injury, SI),并发现轴突内钙离子浓度([Ca2+]i)的升高是导致轴突继发性变性及断裂的基础[4-5]。对于DAI后的轴突内[Ca2+]i升高主要认为是通过电压依赖的钙通道[6]、谷氨酸受体[7-9]及膜通透性改变[10]介导的,对于内质网(endoplasmic reticulum, ER)这一重要的参与调节细胞内钙稳态的细胞器在DAI介导的钙升高过程中是否发挥着作用仍未被阐明。近年来的研究证实,DAI后细胞内钙释放参与了轴突早期的[Ca2+]i升高,但是并未揭示导致这种钙释放的亚细胞器来源[11]。同时,亦有研究表明在其他类型的轴突损伤、变性疾病中来自ER的钙离子释放参与介导了轴突的变性过程[12]。因此,本实验采用细胞牵拉装置对原代培养的神经元轴突进行牵拉致伤建立体外DAI模型,通过钙成像技术检测轴突内[Ca2+]i的变化情况,同时利用改变细胞外液钙离子(无钙细胞外液及含钙细胞外液)以阐明牵拉损伤后引起轴突[Ca2+]i变化的钙来源,进一步通过咖啡因促进ER钙排空的方法判断牵拉损伤后ER内钙离子存量,从而揭示轴突牵拉损伤与ER钙释放的关系。
1 材料与方法
1.1 主要试剂与仪器 免疫细胞化学所用一抗:小鼠抗MAP2抗体(No.M4403,美国Sigma);兔抗β-tubulin抗体(No.T3526,美国Sigma)。荧光二抗:山羊抗小鼠Alexa Flour 488 IgG(No.A-10680,美国Molecular Probe);山羊抗兔Alexa Flour 594 IgG(No.A-11037,美国Molecular Probes)。免疫细胞化学所用的其他主要试剂:山羊血清(No.G9023,美国Sigma);Triton X-100(No.T8532,美国Sigma);胎牛血清蛋白(bovine serum albumin,BSA,No.A9418,美国Sigma);抗荧光猝灭剂(含DAPI,No.P10144,美国Molecular Probes)。ER Tracker Red(No.E34250,美国Molecular Probes);Fluo-4 AM(F-23917,美国Molecular Probes);本实验使用的仪器均由悉尼大学医学院Bosche Institute提供,共聚焦显微镜(型号:LSM 510 META, 德国ZEISS),倒置相差显微镜(型号:CKX31/CKX41,日本Olympus),普通光学显微镜(No.DM2700 M 德国Leica)。可牵拉硅胶培养皿(STREX,日本B-Bridge,图1),牵拉装置(No.ST-140-04,日本B-Bridge,图2)。

图1 不同规格的可牵拉硅胶培养皿实物图
Fig.1 Stretchable silicon chambers of various sizes
1.2 神经元原代培养 健康孕17~19 d小鼠,由悉尼大学医学院Medical Foundation下属的动物饲养中心提供,动物使用通过澳大利亚动物伦理委员会授权(项目名称:The role of Store-operated Ca2+entry in Traumatic brain injury;项目编号:5916)。将小鼠置于700 mL/L乙醇中15 s后,无菌条件下剖腹取出胎鼠。断头取脑,放入预冷的含糖的D-Hanks平衡盐液中。在解剖镜下仔细分离去除脑膜及血管,分离出大脑皮质并剪碎成1 mm×1 mm×1 mm大小。组织块以1.25 g/L的胰蛋白酶消化(37 ℃,15 min,Sigma)并轻吹打,制成密度为5×105/mL的单细胞悬液,接种于涂有多聚-L-赖氨酸(Sigma)包被的特制的硅胶培养皿中。在神经元接种前于硅胶培养皿中央放置一条硅胶带(2 mm×20 mm),接种后24 h,待细胞贴壁后移除,从而使得中央区域为轴突聚集区。加完全培养基后放入37 ℃的培养箱内进行培养。于培养2 d时加入5 μmol/L的阿糖胞苷以抑制非神经细胞的过度增殖。作用24 h后,更换完全培养基,以后每2~3 d换液1次,每次更换一半培养基。完全培养基由Neurobasal(Gibco)+B27(Gibco)+谷氨酰胺(Gibco)以及青霉素、链霉素(Sigma)各100 U/mL组成。细胞培养7~12 d后待用。
1.3 神经元纯度及细胞活率的计算 采用免疫细胞化学染色的方法,使用微管相关蛋白2(microtubulin associate protein 2, MAP2)标记神经元的细胞骨架结构,β-微管蛋白(β-tubulin)标记全部细胞的细胞骨架结构,以及DAPI标记全部细胞的细胞核。预热的40 g/L多聚甲醛室温固定细胞10 min,用含1 mL/L Triton X-100的PBS溶液室温下通透细胞10 min。含100 mL/L山羊血清的PBS溶液在室温下抗原封闭1 h后,加入含10 mL/L BSA配制的一抗,其中MAP2 1∶2 000稀释,β-tubulin 1∶1 000稀释,4 ℃过夜。PBS洗涤后加入含10 g/L BSA的二抗室温孵育1 h,洗涤后避光晾干。加入含DAPI的防荧光淬灭剂后封片,干燥后置于4 ℃冰箱待用。共聚焦显微镜在10×放大倍数下随机采集图像,每个样本随机选取5个视野,使用Image J(NIH,美国)软件对细胞计数分析。神经元纯度(%)=MAP2阳性细胞数/(MAP2阳性细胞数+β-tubulin阳性细胞数)
用锥虫蓝(台盼蓝)对细胞进行活性分析。40 g/L台盼蓝室温孵育细胞2 min,然后在普通光学显微镜下观察计数。活率计算仍采用10×放大倍数下随机选择的5个视野进行。细胞活率(%)=台盼蓝阴性细胞数/(台盼蓝阴性+台盼蓝阳性细胞数)
1.4 DAI细胞牵拉模型建立及分组 用细胞牵拉装置建立DAI细胞模型[13-15]。将含有经培养7~12 d神经元的硅胶培养皿固定于牵拉装置上(图2),设置牵拉长度为初始长度的120%,牵拉时间为500 ms,牵拉次数为1次,牵拉方向为1维单轴牵拉。损伤组神经元轴突行上述的一维牵拉损伤,对照组神经元不行牵拉损伤。其中损伤组中根据灌流人工脑脊液(artificial cerebral spinal fluid, ACSF)含钙与否又分为损伤+含钙ACSF及损伤+无钙ACSF两个亚组。

图2 可牵拉硅胶培养皿置于牵拉装置实物图
Fig.2 A stretchable chamber inserted in the stretch device
1.5 钙成像及ER Tracker Red共聚焦成像 用ACSF轻柔洗涤1~2次,加入含200 g/L聚醚F-127终浓度为 2 μmol/L的Fluo-4 AM,37 ℃避光孵育30 min。用ACSF 37 ℃洗涤20 min后,置于激光共聚焦显微镜上观察检测。含钙ACSF配方:(137 mmol/L NaCl,5 mmol/L KCl,5.6 mmol/L glucose,20 mmol/L HEPES,0.6 mmol/L KH2PO4,0.5 mmol/L Na2HPO4,2.0 mmol/L CaCl2,1.0 mmol/L MgCl2)。无钙ACSF:(137 mmol/L NaCl,5 mmol/L KCl,5.6 mmol/L glucose,20 mmol/L HEPES,0.6 mmol/L KH2PO4,0.5 mmol/L Na2HPO4, 3.0 mmol/L MgCl2,1 mmol/L EGTA)。激光共聚焦显微镜使用参数为:激发光488 nm,出射光505 nm,扫描速度:0.075 μm/像素,512像素/行,间隔2 ms。并用同一细胞在未牵拉情况下的荧光信号为基线。在牵拉损伤后2、5、10、30 min共4个时间点连续10次扫描,计算10次扫描信号强度的均值为钙离子相对浓度。计算公式为:△F=[(F-Fb)-(F0-F0b)]/(F0-F0b),其中F0为损伤前荧光信号值,Fb为背景荧光值,F0b为对照组背景荧光值,F为损伤后荧光信号值。
ER Tracker Red共聚焦成像过程与上述过程相似。加入用含20 μmol/L ER Tracker Red的完全培养基,孵育3 min后用完全培养基洗涤2次,置于共聚焦显微镜下观察,测量参数及计算方法同上。
1.6 统计学分析 数据处理采用SPSS 17.0软件进行统计学分析。实验数据以均值±标准误表示,各时间点上实验组与对照组的比较采用单因素方差分析(ANOVA),两两比较采用LSD-t检验,以P<0.05为差异有统计学意义。
2 结 果
2.1 原代培养的神经元生长状态的形态学观察、纯度及活率鉴定 倒置相差显微镜下直接观察可见,体外培养7~12 d神经元之间产生相互连接,轴突间形成十分密集的轴突网,标志着神经元已经进入V期(stage V)成熟阶段(图3A)。荧光双标检测神经元纯度结果显示:所培养细胞中神经元所占比例为(91.32±4.63)%(图3B~D)。台盼蓝染色结果显示神经元活率为(97.68±1.21)%。
图3 原代培养7~12 d神经元形态学观察及纯度鉴定
Fig.3 Morphological observation and purity identification of primary cultured neurons
A:倒置相差显微镜下观察;B:MAP2免疫细胞化学荧光染色(绿色荧光);C:β-tubulin 免疫细胞化学荧光染色(红色荧光);D:MAP2(绿色荧光)+β-tubulin(红色荧光)+DAPI(蓝)荧光合成图;图中白色箭头所示为非神经元细胞;标尺=50 μm。
2.2 牵拉损伤后轴突内钙[Ca2+]i浓度测定与轴突的形态变化 行牵拉致伤后的神经元轴突即刻产生屈曲的形态学变化;在牵拉损伤后,轴突内[Ca2+]i逐步升高(图4A~D)。损伤后2 min测量轴突内[Ca2+]i已明显升高,与损伤前比较,差异有统计学意义(P<0.05),在随后的30 min内,轴突[Ca2+]i呈阶梯状升高(图4G)。牵拉损伤后轴突变性也伴随着轴突[Ca2+]i的升高而发生,随着轴突内[Ca2+]i升高,轴突内的局部区域具有较高的[Ca2+]i上升部位出现了肿胀,与肿胀部位前后连接的轴突部分出现皱缩,形成了“串珠样”结构。随后,肿胀程度进一步加重,进而出现轴突的继发性断裂(图4E、4F)。

图4 牵拉损伤后单个轴突的钙[Ca2+]i浓度变化
Fig.4 Analysis of single axonal calcium concentration after stretch-induced injury
A~D:Fluo-4荧光共聚焦图,为牵拉损伤前及牵拉损伤后2、10、30 min;E~F:共聚焦显微镜透射光成像,分别为牵拉损伤前、牵拉损伤后30 min;G:各时间点的轴突[Ca2+]i结果分析。损伤组与对照组比较,*P<0.05;标尺=50 μm。
2.3 含钙与无钙细胞外液条件下细胞内[Ca2+]i浓度的测定 将细胞外液换成不含钙离子的ACSF,并在牵拉损伤前,预先将细胞在无钙ACSF下孵育15 min,使[Ca2+]i稳定后进一步行牵拉损伤及钙测量。细胞内[Ca2+]i浓度结果(图5)显示,牵拉损伤后2~30 min,细胞处于无钙的细胞外液环境中,轴突内[Ca2+]i浓度仍然在牵拉损伤后升高,与对照组比较,差异有统计学意义(P<0.05)。在损伤后2 min,不含钙细胞外液条件下的轴突内[Ca2+]i升高与含钙细胞外液条件下轴突内[Ca2+]i升高程度无明显区别(P>0.05),但5~30 min的含钙ACSF条件下的[Ca2+]i升高程度明显高于无钙ACSF条件下的[Ca2+]i升高程度,各组间比较,差异有统计学意义(P值均<0.05)。
2.4 ER Tracker Red对轴突部位共聚焦成像 结果显示ER存在于轴突中(图6A);同时发现ER的荧光强度与轴突的管腔粗细直接相关,直径较大的轴突具有更多的ER数量,反之相对直径较细的轴突则仅具有较少的ER数量(图6A)。牵拉损伤后,随着轴突变性的发生,ER在轴突肿胀部位发生聚集(图6B)。
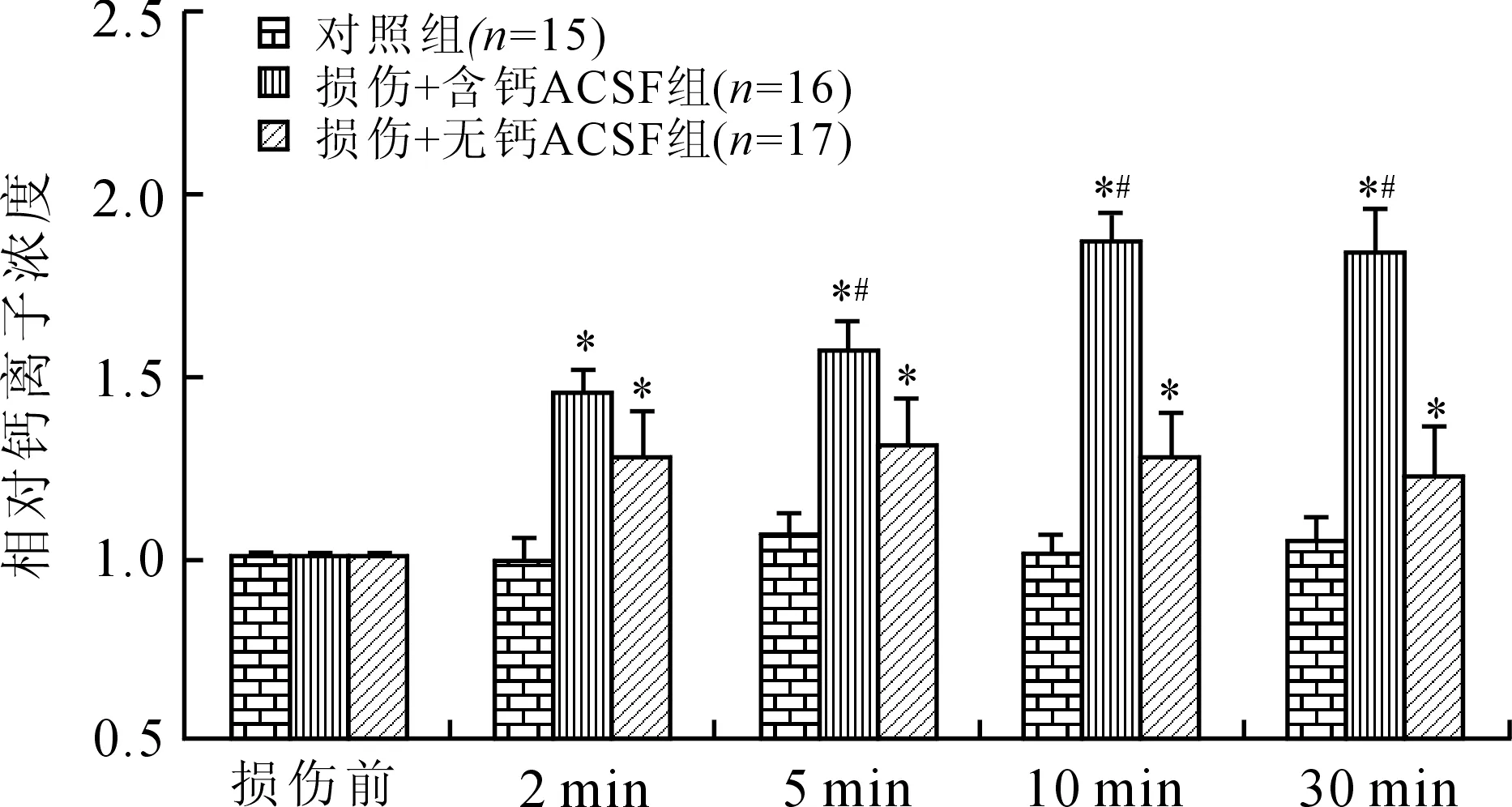
图5 不同组间轴突[Ca2+]i浓度变化
Fig.5 Analysis of axonal [Ca2+]iconcentration in different groups与对照组比较,*P<0.05;与损伤+无钙ACSF组比较,#P<0.05。
2.5 咖啡因诱导的钙反应 对照组轴突(未进行牵拉致伤),在20 mmol/L咖啡因作用下,可激活一个快速、剧烈的[Ca2+]i升高(图7A~C)。而对于牵拉损伤的轴突,在损伤后2 min,进行同样的20 mmol/L咖啡因干预,其激活的钙浓度升高相对较低(图7D~F),两组比较差异具有统计学意义(图7G,P<0.05)。
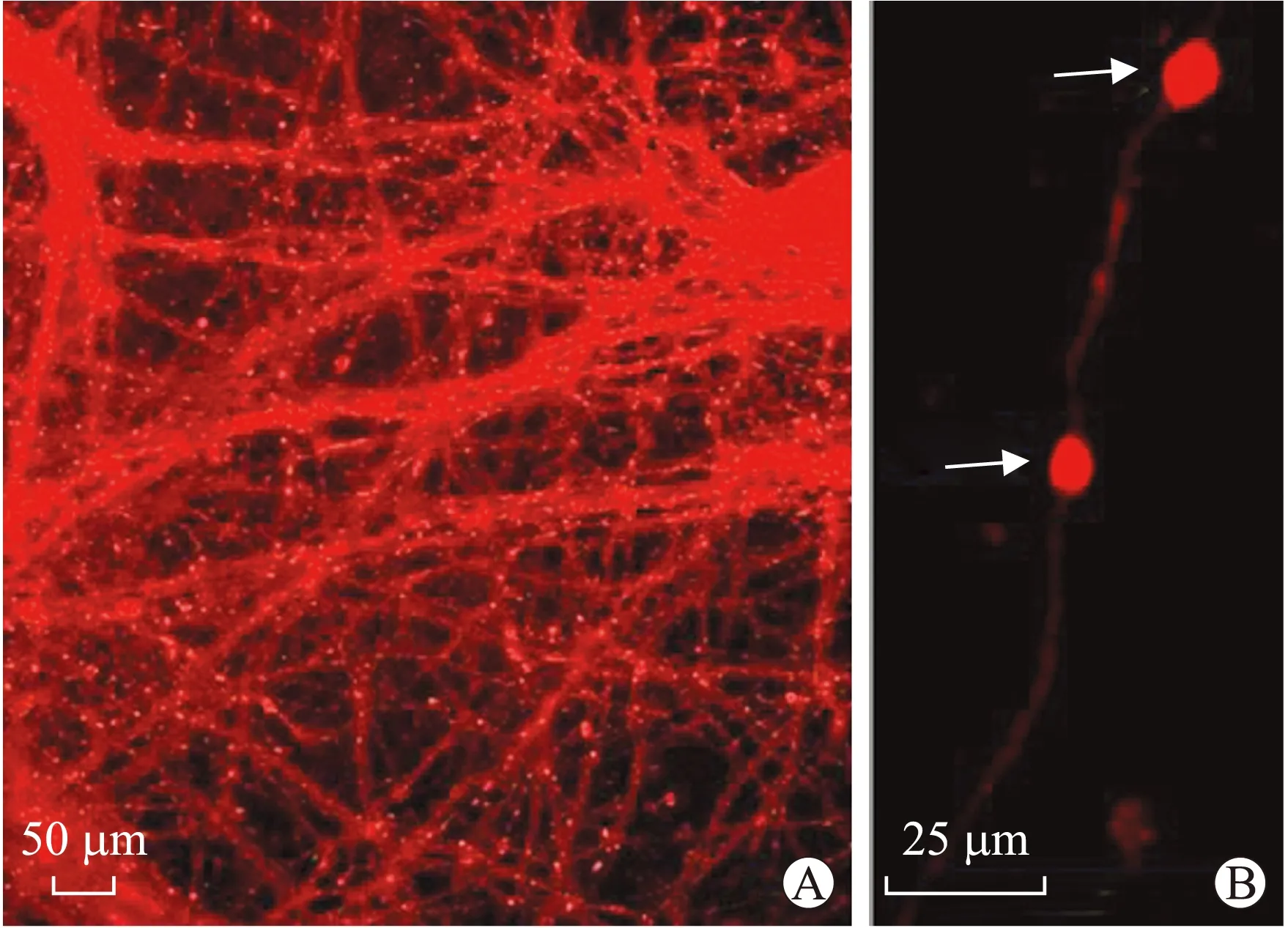
图6 ER Tracker Red 共聚焦成像
Fig.6 Confocal images of ER Tracker Red
A:轴突部位ER Tracker Red 标记的ER(红色荧光所示);B:牵拉损伤后,ER在轴突变性肿胀部位聚集(箭头所示)。

图7 咖啡因诱导的钙反应
Fig.7 Caffeine-induced calcium response
A~C:分别为对照组轴突钙离子基线浓度、2 min后钙离子浓度及加入20 mmol/L咖啡因后的钙离子浓度;D~F:分别为损伤前轴突钙离子基线浓度、损伤后2 min钙离子浓度及加入20 mmol/L咖啡因后的钙离子浓度;G:咖啡因诱导的轴突[Ca2+]i增加幅度(分别与各自基线荧光强度进行比较的倍数)。损伤组与对照组比较,*P<0.05;标尺=50 μm。
3 讨 论
本实验使用细胞牵拉装置建立了体外的神经元轴突牵拉损伤模型,实验中所使用的细胞牵拉装置已被广泛使用于多种其他类型细胞的牵拉损伤的研究中,如平滑肌细胞[14]、心肌细胞[16]、血管内皮细胞[17]、成骨细胞[18]等。本实验通过使用该装置对神经元细胞进行牵拉致伤,损伤后的轴突表现牵拉所致的屈曲,但并未在损伤后立刻产生神经元轴突的断裂(图4A、B),同时牵拉未导致细胞及轴突的脱壁现象;随后,被牵拉后的轴突表现出一个进行性的肿胀、变性及断裂过程,符合DAI后轴突的病理学改变(图4E、F);同时,牵拉后的轴突内钙离子浓度明显升高(图4C、D),这点与DAI后的病理生理机制一致[19],这些证据充分说明了本实验建立的模型是可靠的。与其他同类型的DAI细胞模型相比较,该模型由于使用特制硅胶培养皿进行牵拉致伤,可以一次同时牵拉多个神经元及其轴突,以满足如免疫细胞化学、PCR及Western blot 等需要较多细胞数量完成的实验研究,同时与DAI动物模型比较,该模型能更好对细胞结构进行损伤后的快速观察及功能测量。
由于大量研究已经证实,DAI后导致轴突继发性变性及断裂的基础是轴突内钙离子浓度([Ca2+]i)升高,但对其升高的来源目前仍然不十分清楚。本实验分别采用了含钙的细胞外液及无钙的细胞外液,将细胞分别置于这两种细胞外液中行牵拉损伤。结果发现在含钙细胞外液中,牵拉后的轴突钙离子浓度明显升高,并在10 min内呈进行性升高的趋势。在无钙的细胞外液中,牵拉损伤并未完全抑制轴突[Ca2+]i升高,说明了细胞内的钙释放参与了牵拉损伤后的[Ca2+]i升高;另外,在牵拉后的2 min时间点,无钙细胞外液及含钙细胞外液条件下的牵拉损伤所引发的[Ca2+]i升高程度无明显差异,说明损伤后2 min左右细胞内钙释放是占主导作用的,而5~30 min阶段的[Ca2+]i升高则是由细胞内钙释放和细胞外钙内流共同参与的,这与STAAL[11]及IWATA等[4]报道的实验结果一致。
一直以来,对于DAI后钙离子浓度升高的机制研究大都是围绕细胞外钙离子向细胞内的转移这一途径进行阐明,而对ER与外伤乃至其他类型神经元损伤、变性疾病的联系知之甚少。由于ER是神经元内最大的细胞内钙库,在维持细胞内钙稳态中发挥着重要作用[20]。目前,对于其他中枢神经系统疾病如阿尔茨海默病[21]及亨廷顿病[22]等,ER的钙释放都被逐渐证明与疾病的发生及演进过程关系密切。但在脑外伤方面,ER的钙释放是否参与其中仍未被探明。本实验首先证实了ER存在于神经元的轴突结构中,进一步通过咖啡因的使用来判断牵拉损伤后细胞内钙释放的来源是否为ER,结果显示,对照组轴突(未进行牵拉致伤)在20 mmol/L咖啡因的作用下,可激活一个快速、剧烈的[Ca2+]i升高(图7A~C),这种轴突内[Ca2+]i的升高来源于ER的钙释放作用,因而间接说明了ER管腔内钙离子的存量。而对于牵拉损伤的轴突,在损伤后2 min,进行同样的20 mmol/L咖啡因干预,其激活的钙离子浓度升高程度明显低于对照组,说明牵拉损伤后轴突细胞内钙释放的细胞器来源是ER。本实验研究结果证实了ER在DAI后的早期通过释放钙离子至轴突胞质,参与了DAI后早期(2~30 min)轴突钙超载的形成,这对轴突牵拉损伤乃至临床上DAI的发病机制是一种新的认识。
由于ER作为神经元内的最重要的钙库,提供了一个巨大的细胞内钙存量(ER内钙离子浓度约为胞浆游离钙离子浓度的10的4~5次方倍),在位于ER上的三磷酸肌醇受体 (Inositol 1,4,5-triphosphate receptors, IP3Rs)或雷诺啶受体(Ryanodine receptor, RyRs )被激活后ER内钙离子可以释放至胞质[23]。在正常生理状态下,神经元ER内维持一定浓度的IP3分子,维持神经元突触信号传递所需的钙波(Calcium wave)形成所需的ER钙离子释放[24]。但在理化因素刺激下,一些细胞外配体可以增加ER和胞膜对钙细胞表面受体活化G蛋白,活化的G蛋白水解磷脂酶C(PLC)从而产生大量IP3。IP3与ER表面的IP3R结合,使得ER钙离子释放[25]。本实验中用到的咖啡因,正是RyR受体激动剂的一种,较高浓度的咖啡因可直接激活RyR受体,使得ER内钙离子迅速释放。本研究证明了DAI后ER钙释放的存在,但并未探明所可能涉及的分子机制,推测可能牵拉损伤作为一个刺激信号,将快速激活细胞内的IP3信号分子的聚集,这种聚集可能是由于机械牵拉作用对膜上G蛋白活化,继而水解PLC后产生。另一种可能是牵拉损伤将激活牵拉相关通道(stretch activated channels, SAC),SAC被证实与RyR受体在功能上有藕联的关系。即牵拉损伤激活SAC,SAC将激活RyR继而使得ER钙释放,但具体SAC与RyR藕联通过何种分子机制,目前还不清楚[26]。
另外,本实验发现牵拉损伤后ER在轴突变性肿胀部位聚集。目前对于该病理变化未见其他文献报道。推测导致这种病理改变的原因可能与牵拉损伤后引起的轴浆运输中断有关,轴浆运输的物质包括各种细胞器结构沿着微管自轴突近端运输到轴突末梢,牵拉损伤将导致微管的破坏,从而中断轴浆运输过程,这可能是ER在变性的轴突区域聚集的原因[27]。
总之,我们研究结果证实了DAI后的轴突钙离子浓度升高不仅由细胞外钙内流引起,细胞内钙释放同样参与其中,ER是引起这种DAI后轴突内钙释放的细胞器来源,ER的钙释放参与了轴突牵拉损伤后的[Ca2+]i浓度升高,但抑制这一病理生理过程是否可以减轻损伤后的轴突继发性变性或断裂,本实验并未阐明。未来对于ER钙释放在DAI乃至其他类型脑外伤疾病中的作用值得更为深入、全面的研究。
[1] CHELLY H, CHAARI A, DAOUD E, et al. Diffuse axonal injury in patients with head injuries: an epidemiologic and prognosis study of 124 cases[J]. J Trauma, 2011, 71(4):838-846.
[2] 宋锦宁,刘守勋,戈治理,等. 脑弥漫性轴索损伤的特点及临床诊断[J]. 中国神经精神疾病杂志, 1997, 23(3):141-144, 197.
[3] SMITH DH, MEANEY DF, SHULL WH. Diffuse axonal injury in head trauma[J]. J Head Trauma Rehabil, 2003, 18(4):307-316.
[4] IWATA A, PK S, JA W. Traumatic axonal injury induces proteolytic cleavage of the voltage-gated sodium channels modulated by tetrodotoxin and protease inhibitors[J]. J Neurosci, 2004, 24(19):4605-4613.
[5] 李宇,宋锦宁,闫文涛. DAI后神经元钙离子超载机制的研究现状及进展[J]. 中华神经医学杂志, 2012, 11(5):527-530.
[6] WOLF JA, STYS PK, LUSARDI T, et al. Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels[J]. J Neurosci, 2001, 21(6):1923-1930.
[7] PARK E, BELL JD, BAKER AJ. Traumatic brain injury: can the consequences be stopped?[J]. CMAJ, 2008, 178(9):1163-1170.
[8] FEI Z, ZHANG X, BAI HM, et al. Posttraumatic secondary brain insults exacerbates neuronal injury by altering metabotropic glutamate receptors[J]. BMC Neurosci, 2007, 8:96.
[9] SPAETHLING J, LE L, MEANEY DF. NMDA receptor mediated phosphorylation of GluR1 subunits contributes to the appearance of calcium-permeable AMPA receptors after mechanical stretch injury[J]. Neurobiol Dis, 2012, 46(3):646-654.
[10] KILINC D, GALLO G, BARBEE KA. Mechanically-induced membrane poration causes axonal beading and localized cytoskeletal damage[J]. Exp Neurol, 2008, 212(2):422-430.
[11] STAAL JA, DICKSON TC, GASPERINI R, et al. Initial calcium release from intracellular stores followed by calcium dysregulation is linked to secondary axotomy following transient axonal stretch injury[J]. J Neurochem, 2010, 112(5):1147-1155.
[12] GEMES G, RIGAUD M, WEYKER PD, et al. Depletion of calcium stores in injured sensory neurons: anatomic and functional correlates[J]. Anesthesiology, 2009, 111(2):393-405.
[13] WHITEHEAD NP, STREAMER M, LUSAMBILI LI, et al. Streptomycin reduces stretch-induced membrane permeability in muscles from mdx mice[J]. Neuromuscul Disord, 2006, 16(12):845-854.
[14] ITO S, MAJUMDAR A, KUME H, et al. Viscoelastic and dynamic nonlinear properties of airway smooth muscle tissue: roles of mechanical force and the cytoskeleton[J]. Am J Physiol Lung Cell Mol Physiol, 2006, 290(6):L1227-L1237.
[15] AMMA H, NARUSE K, ISHIGURO N, et al. Involvement of reactive oxygen species in cyclic stretch-induced NF-kappaB activation in human fibroblast cells[J]. Br J Pharmacol, 2005, 145(3):364-373.
[16] MATSUDA T, FUJIO Y, NARIAI T, et al. N-cadherin signals through Rac1 determine the localization of connexin 43 in cardiac myocytes[J]. J Mol Cell Cardiol, 2006, 40(4):495-502.
[17] KAWAI M, NARUSE K, KOMATSU S, et al. Mechanical stress-dependent secretion of interleukin 6 by endothelial cells after portal vein embolization: clinical and experimental studies[J]. J Hepatol, 2002, 37(2):240-246.
[18] DANCIU TE, ADAM RM, NARUSE K, et al. Calcium regulates the PI3K-Akt pathway in stretched osteoblasts[J]. FEBS Lett, 2003, 536(1-3):193-197.
[19] LI XY, LI J, FENG DF, et al. Diffuse axonal injury induced by simultaneous moderate linear and angular head accelerations in rats[J]. Neuroscience, 2010, 169:357-369.
[20] VERKHRATSKY A. Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons[J]. Physiol Rev, 2005, 85(1):201-279.
[21] WU J, SHIH HP, VIGONT V, et al. Neuronal store-operated calcium entry pathway as a novel therapeutic target for Huntington's disease treatment[J]. Chem Biol, 2011, 18(6):777-793.
[22] ZEIGER W, VETRIVEL KS, BUGGIA-PREVOT V, et al. Ca2+influx through store-operated Ca2+channels reduces Alzheimer disease beta-amyloid peptide secretion[J]. J Biol Chem, 2013, 288(37):26955-26966.
[23] YOSHIOKA M, YAMAZAKI Y, FUJII S, et al. Intracellular calcium ion dynamics involved in long-term potentiation in hippocampal CA1 neurons in mice lacking the IP3 type 1 receptor[J]. Neurosci Res, 2010, 67(2):149-155.
[24] ROSS WN. Understanding calcium waves and sparks in central neurons[J]. Nat Rev Neurosci, 2012, 13(3):157-168.
[25] RUIZ A, MATUTE C, ALBERDI E. Endoplasmic reticulum Ca2+release through ryanodine and IP3 receptors contributes to neuronal excitotoxicity[J]. Cell Calcium, 2009, 46(4):273-281.
[26] SAVINEAU JP, GILBERT G, DUCRET T, et al. Coupling between stretch-activated channels and ryanodine receptors in vacsular smooth muscle cells[J]. The FASEB J, 2013, 27:922.
[27] TANG-SCHOMER MD, JOHNSON VE, BAAS PW, et al. Partial interruption of axonal transport due to microtubule breakage accounts for the formation of periodic varicosities after traumatic axonal injury[J]. Exp Neurol, 2012, 233(1): 364-372.
(编辑 国 荣)
Endoplasmic reticulum calcium release contributes to the elevatedintra-axonal calcium concentration after diffuse axonal injury
LI Yu, SONG Jin-ning, ZHANG Ming, AN Ji-yang, SUN Peng,LI Dan-dong, MA Xu-dong, ZHAO Ya-hui
(Department of Neurosurgery, the First Affiliated Hospital, Medical School ofXi’an Jiaotong University, Xi’an 710061, China)
Objective To investigate whether calcium release from endoplasmic reticulum (ER) contributes to the elevated axonal calcium concentration in the early stage of diffuse axonal injury (DAI) and discuss the possible molecular mechanisms of this ER calcium release. Methods We used primary cultured neurons of day 7-12 mice to establishinvitroDAI model by axonal stretch-induced injury. ER Tracker Red was used for detecting the ER distribution in axons. In addition, Fluo-4 AM was used to measure the change of axonal calcium concentration after axonal stretch injury. Through caffeine application, calcium stock in ER lumen was investigated indirectly. Results The data of ER Tracker Red fluorescence images demonstrated that ER was present in the axons. And after stretch-induced injury, more ER organelles accumulated in the swollen axonal structures. Axons exhibited a sharp intra-axonal calcium increase after stretch-induced injury. And this increased intra-axonal calcium was associated with axonal swelling, compared with the control group (P<0.05). Zero calcium extracellular buffer did not blunt the intra-axonal calcium increase caused by stretch at 2 min post injury, compared with the normal calcium extracellular buffer group (P>0.05). However, at the other time points post injury, zero calcium extracellular buffer significantly depressed the intra-axonal calcium increase, compared with the normal calcium extracellular buffer group (P>0.05). Furthermore, 20 mmol/L caffeine triggered a huge and quick calcium increase in the axons of the control group. But for axons which had been stretched in advance, 20 mmol/L caffeine only triggered a relatively smaller calcium increase, compared with the control group (P<0.05). Conclusion Extracellular calcium influx is not the only mechanism which results in the elevated concentration of axonal intracellular calcium. Intracellular calcium release also plays a role. And ER is the source of this intracellular calcium release.
traumatic brain injury; diffuse axonal injury; calcium; endoplasmic reticulum
2014-04-21
2014-06-28
国家自然科学基金资助项目(No.30471774);教育部新世纪优秀人才支持计划资助项目(No.NCET-05-0831);陕西省自然科学基金资助项目(No.2003C1-16) Supported by the National Natural Science Foundation of China (No.30471774), the New-Century Excellent Talents Program of Ministry of Education (No.NCET-05-0831), and the Natural Science Foundation of Shaanxi Province (No.2003C1-16)
宋锦宁,博士,教授,主任医师,博士生导师. E-mail: jinnings@126.com
李宇(1986-),男(汉族),博士研究生在读. 研究方向:脑外伤的基础及临床. E-mail: liyuwood@gmail.com
时间:2014-09-16 11∶34 网络出版地址:http://www.cnki.net/kcms/detail/61.1399.R.20140916.1134.005.html
R651.1+5
A
10.7652/jdyxb201406002
猜你喜欢
浙江临床医学(2021年12期)2021-11-29
中华养生保健(2020年10期)2021-01-18
中南民族大学学报(自然科学版)(2019年3期)2019-10-11
保健与生活(2019年23期)2019-09-10
体育科学(2018年11期)2018-12-07
中学生理科应试(2017年6期)2017-09-27
军事体育学报(2016年4期)2016-05-11
中华神经创伤外科电子杂志(2015年1期)2015-01-21
西南医科大学学报(2014年6期)2014-03-20
中南民族大学学报(自然科学版)(2014年2期)2014-01-22
